


28 травня, 2020
Сучасні підходи до діагностики та лікування кардіоміопатій: що нового?
У грудні 2019 року в м. Києві відбулася підсумкова науково-практична конференція Київського кардіологічного товариства, організована ГО «Всеукраїнська асоціація кардіологів України».
 Керівник Експертного консультативно-діагностичного та лікувального центру міокардиту і кардіоміопатій ДУ «Національний науковий центр «Інститут кардіології ім. М.Д. Стражеска» НАМН України» (м. Київ), доктор медичних наук, професор Олена Геннадіївна Несукай розглянула питання механізмів формування та підходи до лікування серцевої недостатності (СН) при кардіоміопатіях (КМП).
Керівник Експертного консультативно-діагностичного та лікувального центру міокардиту і кардіоміопатій ДУ «Національний науковий центр «Інститут кардіології ім. М.Д. Стражеска» НАМН України» (м. Київ), доктор медичних наук, професор Олена Геннадіївна Несукай розглянула питання механізмів формування та підходи до лікування серцевої недостатності (СН) при кардіоміопатіях (КМП).
– Термін «кардіоміопатія» об’єднує гетерогенну групу хвороб серцевого м’яза, що включає дилатаційну (ДКМП), гіпертрофічну (ГКМП), рестриктивну (РКМП), аритмогенну правого шлуночка та некласифіковану КМП (некомпактну), котрі рано чи пізно маніфестують СН. Розмаїття етіологічних чинників, патофізіологічних механізмів і фенотипу зумовлює особливості клінічної картини КМП і відсутність системних оглядів, присвячених особливостям відповіді на їх лікування. На сучасному етапі останні досягнення в розумінні механізмів розвитку СН при КМП, нові розробки, пов’язані з етіологією (замісна терапія ферментами, стабілізатори транстиретину, імунотерапія), суттєво впливають на стратегію лікування та покращення його результатів. У 2019 році Асоціація серцевої недостатності Європейського товариства кардіологів оприлюднила позиційний документ «Серцева недостатність при кардіоміопатіях» (Seferović P. et al., 2019), у якому розглянуто питання епідеміології, механізми розвитку СН, а також останні досягнення в менеджменті хворих із різними КМП.
Дилатаційна кардіоміопатія
ДКМП характеризується збільшенням об’єму шлуночків серця та зниженням їхньої систолічної функції в умовах відсутності встановленого етіологічного чинника чи значущого ураження коронарних артерій. Якщо раніше ДКМП вважалася хворобою нез’ясованої етіології, то нині відомо, що основою для розвитку дисфункції серця при ДКМП є як генетичні передумови, так і прямий ушкоджувальний вплив інфекції, токсичних агентів, ендокринних, метаболічних, імуноопосередкованих та інших процесів, а також перипартальна КМП (рис. 1). Сьогодні відомо понад 60 генів, які кодують білки саркомерів, цитоскелет, ядерну оболонку, сакролему, іонні канали; більшість мутацій мають аутосомно-домінантний тип успадкування.

Рис. 1. Етіологія дилатаційної кардіоміопатії
Примітки: ВІЛ – вірус імунодефіциту людини; СЧВ – системний червоний вовчак; ТЦА – трициклічні антидепресанти; ФВ ЛШ – фракція викиду лівого шлуночка.
У пацієнтів із ДКМП можливі три варіанти перебігу СН: структурне та функціональне відновлення після епізоду СН, виникнення ремісії з покращенням або стабілізацією перебігу СН, а також прогресування до тяжкої СН, що спричиняє смерть або потребу в трансплантації. Нормалізація структури та функції лівого шлуночка (ЛШ) відбувається при застосуванні чинних рекомендацій із лікування СН.
Результати нещодавно проведеного відкритого пілотного рандомізованого дослідження з відміни фармакологічного лікування СН у пацієнтів із ДКМП (TRED-HF) за участю 51 пацієнта з відновленою функцією ЛШ продемонстрували, що припинення лікування СН згідно з рекомендаціями спричиняє рецидив систолічної дисфункції в 40% випадків упродовж 6 міс спостереження (Halliday B. et al., 2019). Ці дані свідчать про необхідність продовження лікування СН навіть за відсутності маніфестації ознак ДКМП. На сучасному етапі етіологічне лікування ДКМП перебуває в стадії розвитку та потребує припинення дії причинного фактора (наприклад, відмова від уживання алкоголю, усунення ендокринних і метаболічних порушень) і поглиблення доказової бази новими результатами клінічних досліджень. У дослідженні IMPROVE-HF із залученням 3994 пацієнтів із СН предикторами функціонального відновлення ЛШ було визначено жіночу стать, неішемічну етіологію СН і відсутність дигоксину в схемах лікування (Wilcox J.E., 2012). До предикторів несприятливого перебігу ДКМП відносять знижену ФВ ЛШ і вищий функціональний клас, виражену мітральну регургітацію, наявність повної блокади лівої ніжки пучка Гіса та підвищеного вмісту натрійуретичного пептиду. Як відомо, хірургічна корекція вираженої мітральної недостатності забезпечує покращення функціональних характеристик і розвиток зворотного ремоделювання ЛШ, що стало поштовхом до оптимізації перкутанних методик пластики мітрального клапана.
Гіпертрофічна кардіоміопатія
ГКМП діагностується в дорослих за умови товщини стінки ЛШ ≥15 мм в одному чи кількох сегментах або ≥13 мм у дорослих, близькі родичі котрих мають встановлену ГКМП, за відсутності хвороб, які характеризуються патологічним перевантаженням ЛШ (клапанні вади серця, артеріальна гіпертензія). У більшості пацієнтів спостерігається асиметрична гіпертрофія ЛШ, у 40-70% хворих на ГКМП діагностується обструктивний варіант, якому притаманне збільшення градієнта тиску на виносному тракті (ВТ) ЛШ до ≥30 мм рт. ст. у стані спокою (близько 25% пацієнтів) або під час фізичного навантаження. У 60% випадків наявність фенотипу ГКМП зумовлена мутаціями генів, які кодують білки саркомерів, а в 5-10% випадків є результатом успадкованих синдромів, нервово-м’язових захворювань і хвороб накопичення (хвороба Андерсона-Фабрі (ХАФ), Помпе, Данона та ін.). Більш ніж у 30% хворих причина розвитку ГКМП залишається нез’ясованою. СН при ГКМП має дві характерні клінічні особливості: в більшості пацієнтів маніфестує як СН зі збереженою ФВ на тлі обструкції ВТ ЛШ; у невеликої кількості хворих на пізніших стадіях захворювання спостерігається СН зі зниженою ФВ ЛШ.
За відсутності обструкції ВТ ЛШ пацієнти переважно безсимптомні. У хворих з обструктивною формою ГКМП тяжкість СН насамперед зумовлена вираженістю перенавантаження ЛШ тиском унаслідок динамічної обструкції ВТ під час систоли. Обструкція порожнини ЛШ на рівні середніх сегментів часто асоціюється з тяжким перебігом СН і високим показником смертності. Іншою важливою причиною розвитку СН у пацієнтів із ГКМП є діастолічна дисфункція серця, що спостерігається в більшості хворих незалежно від наявності обструкції ВТ і характеризується подовженням фази ізометричної релаксації м’язових волокон і порушенням наповнення ЛШ у діастолу. Важливим фактором несприятливого перебігу ГКМП в осіб молодого віку є ранній розвиток фібриляції передсердь. Стратегія лікування СН у пацієнтів із ГКМП об’єднує стандартну терапію СН та етіотропну терапію. Препаратами першої лінії при лікуванні пацієнтів із ГКМП є β-блокатори (ББ) без вазодилатуючих властивостей, які призначаються в комбінації з низькими дозами діуретиків. За наявності протипоказань до ББ альтернативою можуть бути верапаміл або дилтіазем (рис. 2). У симптомних хворих з обструктивною формою ГКМП і збереженою ФВ ЛШ попри прийом максимальних доз ББ доцільним може бути додавання дизопіраміду. До перспективних напрямів у лікуванні ГКМП, розроблених останнім часом, належить використання ранолазину. У пацієнтів із ГКМП дисрегуляція передачі сигналу кальцієвими каналами в кардіоміоциті призводить до розвитку діастолічної дисфункції й аритмогенності, часто передує гіпертрофії ЛШ і появі симптомів СН. Механізмом дії ранолазину є пригнічення саркомерної міграції натрію та підвищення відтоку кальцію за допомогою натрій-кальцієвого обмінника, наслідком чого є зниження аритмогенності та діастолічної дисфункції в пацієнтів із ГКМП. Нещодавно було показано, що пряма модуляція саркомерів при ГКМП має потенціал для нормалізації контрактильності, відновлення лузитропії та, можливо, зменшення симптомів і побічних результатів. У відкритому клінічному дослідженні ІІ фази було показано, що мавакамтен, пероральний малий молекулярний модулятор серцевого міозину, через 12 тиж лікування здатний зменшити обструкцію ВТ ЛШ і покращити працездатність у пацієнтів із симптомною обструктивною ГКМП (Heitner S.B. et al., 2019).
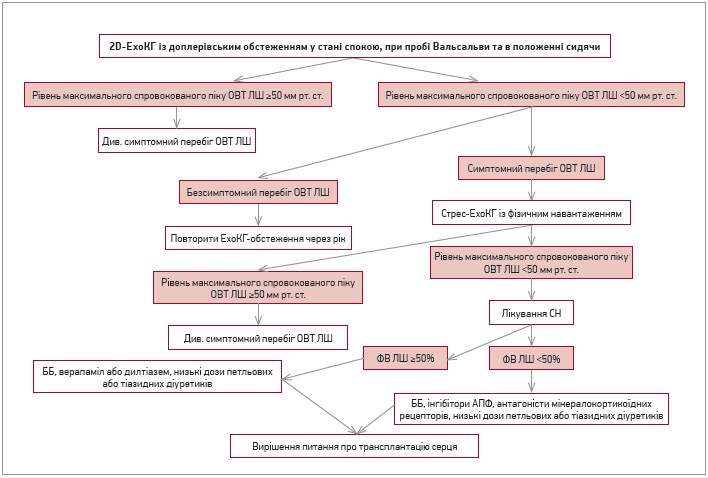
Рис. 2. Тактика ведення пацієнтів із ГКМП
Примітки: ОВТ ЛШ – обструкція вихідного тракту лівого шлуночка; АПФ – ангіотензинперетворювальний фермент.
Для первинної профілактики раптової кардіальної смерті (РКС) і потреби в імплантації кардіовертера-дефібрилятора Європейське товариство кардіологів рекомендує використання спеціальної прогностичної моделі для індивідуалізованої оцінки 5-річного ризику РКС. Для цього оцінюють анамнез, проводять доплер-ехокардіографію (доплер-ЕхоКГ) і 48-годинне амбулаторне моніторування електрокардіограми (ЕКГ). До факторів ризику належать вік, сімейний анамнез РКС, синкопальні стани нез’ясованого генезу, величини ЕхоКГ-показників (градієнт тиску на ВТ ЛШ, максимальна товщина стінки ЛШ, діаметр лівого передсердя) та наявність нестійкої шлуночкової тахікардії.
Рестриктивна кардіоміопатія
РКМП характеризується рестриктивною фізіологією серця, принциповою ознакою котрої є зменшення ударного об’єму на тлі нормальних або зменшених діастолічних і систолічних об’ємів серця. Товщина стінок шлуночків зазвичай не відрізняється від норми, але при хворобах накопичення може спостерігатися потовщення стінок серця. Основною ознакою РКМП є збільшення жорсткості стінок серця за рахунок патологічних змін міокарда та/або ендокарда. РКМП є поліетіологічним захворюванням і може бути ідіопатичною, спадковою та набутою внаслідок інфільтративних і неінфільтративних уражень міокарда, а також виникати на тлі хвороб накопичення. За наявності специфічних етіологічних чинників фенотип захворювання може поєднувати риси ГКМП і РКМП (хвороби Андерсона-Фабрі, Помпе, Данона тощо) чи трансформуватися з фенотипу РКМП у ДКМП при прогресуванні захворювання (гемохроматоз, амілоїдоз та ін.). РКМП є найбільш рідкісним фенотипом серед КМП і в більшості випадків класичним клінічним її проявом є СН зі збереженою ФВ ЛШ, зниження ФВ ЛШ відбувається на пізніх стадіях захворювання й більш характерне для амілоїдозу серця та гемохроматозу. Важливо зазначити, що у хворих віком понад 65 років РКМП як причина розвитку нез’ясованої СН може бути нерозпізнаною. РКМП характеризується високим ризиком розвитку тромбоемболій, порушень ритму та провідності й РКС. Стандартні підходи до лікування РКМП включають рекомендації щодо обмеження вживання рідини та солі (в тому числі у хворих із гіпонатріємією) й обережного використання петльових діуретиків та антагоністів мінералокортикоїдних рецепторів з огляду на те, що великі дози сечогінних препаратів в умовах рестриктивного типу наповнення можуть призводити до гіпотензії внаслідок зменшення об’єму циркулюючої крові. З особливою пересторогою слід використовувати інгібітори АПФ і сартани навіть у невеликих і середніх дозах, оскільки це також може спричиняти надмірне зниження артеріального тиску в пацієнтів із РКМП, а ББ можуть навіть погіршувати перебіг СН, адже невеликий ударний об’єм ЛШ потребує більшої частоти серцевих скорочень для забезпечення адекватної гемодинаміки.
Порушення серцевого ритму зазвичай важко переносяться пацієнтами з РКМП і тому потребують антиаритмічної терапії. Питання щодо доцільності імплантації кардіовертера-дефібрилятора пацієнтам із РКМП зі збереженою ФВ ЛШ донині залишається дискусійним і потребує індивідуалізованого підходу в конкретного хворого з огляду на оцінку аритмогенного ризику, очікувану тривалість життя й інші чинники. На сьогодні недостатньо даних щодо ефективності застосування засобів гемодинамічної підтримки в пацієнтів із РКМП. При встановленні етіологічного чинника РКМП необхідно застосовувати етіотропне лікування.
Хвороба Андерсона-Фабрі
ХАФ – це спадкова хвороба накопичення, що виникає внаслідок порушення синтезу лізосомального ферменту α-галактозидази А, що призводить до накопичення глікосфінголіпідів, переважно глоботріаозилкераміду (Gb3, GL‑3). Ранніми патофізіологічними стадіями захворювання є лізосомне накопичення з подальшим енергетичним і функціональним виснаженням. Надалі виникає компенсаторна гіпертрофія кардіоміоцитів. Урешті, внаслідок прискореного апоптозу клітини міокарда гинуть, розвивається фіброз (Palecek T., 2015). Хвороба маніфестує здебільшого у віковому періоді після 30-35 років, за відсутності лікування тривалість життя істотно зменшується.
Відповідно до Канадських рекомендацій із лікування хвороби Фабрі (2016), для діагностики потрібна наявність щонайменше двох таких критеріїв: товщина стінок ЛШ >12 мм у чоловіків і >11 мм у жінок; гіпертрофія ЛШ на ЕКГ >5 за шкалою Estes; індекс маси міокарда (ІММ) ЛШ при 2D-ЕхоКГ на 20% вищий за вікову норму; річний приріст ІММ ЛШ ≥5 г/м2 (за трьома вимірами впродовж 12 міс); порушення діастолічного наповнення при 2D-ЕхоКГ, діастолічна дисфункція 2-3 ступеня та/або порушення стрейну при спекл-трекінг ЕхоКГ; зміни швидкості поздовжньої та/або радіальної деформації; збільшення лівого передсердя при 2D-ЕхоКГ >40 мм по парастернальній довгій осі (PLAX) або його об’єм >34 мл/м2; порушення ритму та провідності – атріовентрикулярна блокада чи блокада лівої ніжки пучка Гіса, укорочення інтервалу PR, шлуночкові чи передсердні тахіаритмії, синусова брадикардія за відсутності прийому лікарських засобів із негативною хронотропною дією; помірна-тяжка мітральна чи аортальна недостатність; пізнє накопичення гадолінію при магнітно-резонансній томографії (МРТ); вищий за вікову норму рівень NT-proBNP (Sirrs S. et al., 2016).
Червоними прапорцями при підозрі на ХАФ вважають тривалість PR <120 мс і синусову брадикардію на ЕКГ; гіпертрофію сосочкових м’язів при ЕхоКГ; пізнє накопичення гадолінієвого контрасту в нижньо-задньо-латеральній ділянці при МРТ. Натомість виникнення тяжкої гіпертрофії ЛШ в осіб віком до 20 років або низький (<1,5 мВ) вольтаж на ЕКГ дають змогу виключити наявність ХАФ. Морфологічна верифікація можлива після ендоміокардіальної біопсії (Smid B. et al., 2014).
Характерним для ХАФ є зменшення пікового систолічного стрейну в задньо-базально-латеральних сегментах при спекл-трекінг ЕхоКГ. У ході проведення МРТ у зазначених сегментах спостерігається накопичення гадолінію, тоді як позитронно-емісійна томографія виявляє фокусне захоплення 18F-фтордезоксиглюкози (Imbriaco M. et al., 2018). Описано декілька фенотипів ХАФ: ГКМП без обструкції, апікально-септальна ГКМП з обструкцією, концентрична КМП зі зменшенням стрейну в задньо-нижніх відділах, гіпертрабекулярність (некомпактний міокард) верхівки (Renuka J. et al., 2018). Модель прогресування хвороби є такою: зменшення глобального поздовжнього стрейну → втрата радіальної функції → фіброз і накопичення гадолінію → концентрична гіпертрофія ЛШ.
Сьогодні в Україні скринінг ХАФ рекомендовано проводити всім пацієнтам віком >30 років із діагнозом ГКМП або товщиною стінки ЛШ ≥12-13 мм невстановленої етіології, забір крові для безкоштовної генетичної діагностики ХАФ методом сухої краплі проводиться в лабораторії «Діла». Пацієнтів із позитивним результатом тесту слід скерувати до відповідального генетика в обласний медико-генетичний центр, обласний кардіологічний диспансер або ДУ «ННЦ «Інститут кардіології ім. М.Д. Стражеска» НАМН України». Рекомендовано аналіз сімейного анамнезу для визначення ризику в інших членів родини. Після підтвердження діагнозу пацієнт скеровується до Центру орфанних захворювань у Києві для вирішення питань щодо обліку та подальшого лікування мультидисциплінарною комісією. Для лікування ХАФ нині існує досить ефективна замісна ферментна терапія з використанням агалсидази-α, агалсидази-β або мігаластату.
Аритмогенна КМП правого шлуночка – це вроджена патологія внаслідок мутацій десмосом, яка характеризується фіброзно-жировим заміщенням міокарда правого шлуночка, а також ЛШ. Клінічний дебют аритмогенної КМП зазвичай має вигляд порушення ритму, натомість розвиток СН спостерігається в незначної кількості пацієнтів і лише за умови прогресування захворювання. Нещодавно було показано, що відкладання жиру, найімовірніше, зумовлене порушеннями шляху окислених ліпідів низької щільності oxLDL/CD36/PPARγ. Потенційне значення в лікуванні можуть мати дієта та зниження рівня ліпопротеїнів низької щільності (Stadiotti I. et al., 2019).
Отже, сучасний рівень знань про епідеміологію, патофізіологію, клінічний перебіг і перспективні підходи до лікування СН у пацієнтів із КМП дає можливість обрати адекватну тактику ведення цих хворих. Утім, дотепер залишаються невирішеними питання етіотропної та патогенетичної терапії багатьох КМП, що зумовлює необхідність проведення подальших досліджень на основі принципів доказової медицини.
Підготувала Ольга Королюк
Медична газета «Здоров’я України 21 сторіччя» № 7 (476), квітень 2020 р.