


24 липня, 2020
Сучасні погляди на діагностику та лікування множинної мієломи
Частка множинної мієломи складає приблизно 10% у структурі гематологічної захворюваності [1]. На противагу іншим злоякісним новоутворенням, що метастазують у кістки, остеолітичні ураження при множинній мієломі супроводжуються пригніченням чи повним виключенням активності остеобластів [2]. Ураження кісток найкращим чином діагностується з використанням низькодозової комп’ютерної томографії всього тіла, позитронно-емісійної томографії з комп’ютерною томографією або магнітно‑резонансної томографії [3]. Клінічно множинна мієлома часто характеризується анемією, гіперкальціємією, нирковою недостатністю та підвищеним ризиком розвитку інфекційних ускладнень. Екстрамедулярні прояви захворювання на момент діагностики мають приблизно 1-2% хворих, надалі цей показник зростає до 8% [4].
Діагностика множинної мієломи проводиться відповідно до критеріїв, затверджених Міжнародною робочою групою з мієломи [1]. Окрім наявності у кістковому мозку ≥10% клональних плазмоцитів чи підтвердженої за результатами біопсії плазмоцитоми, для встановлення діагнозу необхідна наявність одного або більше з наступних критеріїв:
- ураження органів-мішеней, так звані критерії CRAB:
- hyperСalcemia – гіперкальціємія: концентрація кальцію у сироватці крові більш ніж
на 0,25 ммоль/л (>1 мг/дл) перевищує верхню межу норми, або >2,75 ммоль/л (>11 мг/дл); - Renal insufficiency – ниркова недостатність: кліренс креатиніну становить <40 мл/хв або концентрація креатиніну у сироватці крові >177 мкмоль/л (>2 мг/дл);
- Anemia – анемія: рівень гемоглобіну більше ніж на 2 г/дл нижчий за нижню межу норми або концентрація гемоглобіну <10 г/дл;
- Bone lesions – ураження кісток: одне або більше остеолітичне вогнище ураження за даними радіографічного дослідження скелета, комп’ютерної або позитронно-емісійної томографії з комп’ютерною томографією;
- hyperСalcemia – гіперкальціємія: концентрація кальцію у сироватці крові більш ніж
- наявність у кістковому мозку ≥60% клональних плазмоцитів;
- співвідношення зв’язаних і вільних сироваткових легких ланцюгів ≥100 (при цьому зв’язані вільні легкі ланцюги мають бути ≥100 мг/л);
- більше 1 вогнища ураження кісток розміром ≥5 мм (за даними магнітно-резонансної томографії).
Якщо наявна підозра на множинну мієлому, пацієнту необхідно виконати тест на М-протеїн за допомогою таких методів, як електрофорез білків сироватки крові, сироваткова імунофіксація та визначення у сироватці крові вільних легких ланцюгів. Приблизно 2% пацієнтів з множинною мієломою не мають М-протеїну за результатами всіх вищезазначених досліджень. Визначення М-протеїну проводиться щомісяця під час лікування та через кожні 3-6 місяців після його закінчення [5-9]. Дослідження кісткового мозку на момент первинної діагностики має включати флуоресцентну гібридизацію in situ (а саме: транслокації t(11;14), t(4;14), t(14;16), t(6;14), t(14;20), трисомія і del(17p)). Додаткову прогностичну інформацію може надати генетичне профілювання [10‑11].
Групи ризику та прогноз. Застосування сучасних схем лікування при множинній мієломі дозволяє досягти медіани виживаності близько 6 років [12]. У підгрупі хворих, які підходять для аутологічної трансплантації стовбурових клітин, 4-річна виживаність становить понад 80%, а медіана загальної виживаності – приблизно 8 років [13-14]. Серед пацієнтів віком старше 75 років медіана загальної виживаності менша (близько 5 років) [12]. Протягом останніх 3-5 років були впроваджені моноклональні антитіла й інші лікарські засоби, що дозволило покращити результати лікування множинної мієломи.
Більш точне прогнозування потребує урахування таких факторів, як поширеність пухлинного процесу (стадія), біологічні особливості пухлини (цитогенетичні зміни) та відповідь на терапію [15-16]. За Міжнародною системою стадіювання для мієломи виділяють три стадії [17]:
- I стадія передбачає наявність у пацієнта всіх характеристик із переліку: концентрація сироваткового альбуміну ≥3,5 г/дл, концентрація сироваткового β2-мікроглобуліну <3,5 мг/л, відсутність цитогенетичних варіантів високого ризику та нормальний рівень сироваткової лактатдегідрогенази;
- II стадія включає характеристики, що не підходять ні для I, ні для III стадій;
- III стадія встановлюється за наявності двох характеристик: концентрація сироваткового β2-мікроглобуліну >5,5 мг/л та наявність цитогенетичного варіанту групи високого ризику (t(4;14), t(14;16), або del(17p)) або підвищення рівня сироваткової лактатдегідрогенази.
Лікування. Застосування у хворих із множинною мієломою талідоміду, бортезомібу та леналідоміду дозволило значно покращити результати лікування. Протягом останнього десятиріччя були впроваджені карфілзоміб, помалідомід, панобіностат, іксазоміб, елотузумаб, даратумумаб, ізатуксимаб та селінексор [18‑21]. Основні режими системної терапії при множинній мієломі наведені в таблиці 1. Вибір тактики лікування через стратифікацію на групи ризику для пацієнтів з уперше виявленою симптомною множинною мієломою представлений на рисунку 1. Результати лікування із застосуванням новітніх препаратів наведені в таблиці 2.


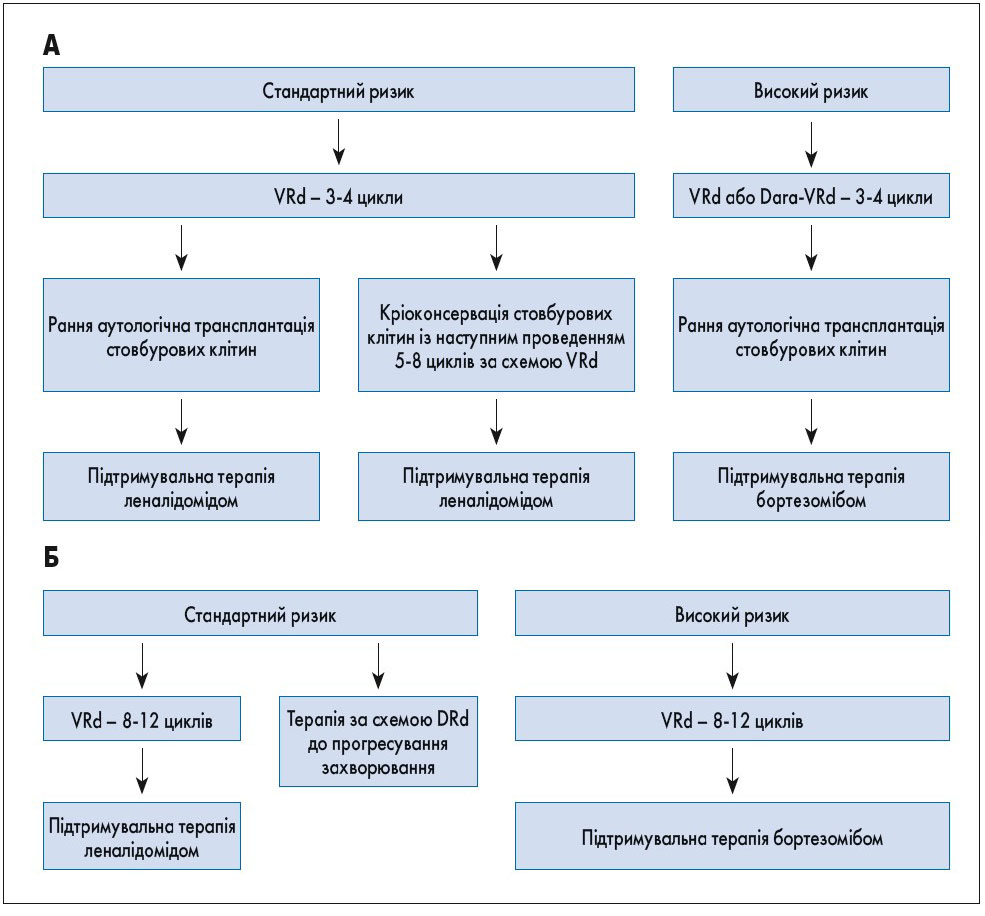 Рис. 1. Тактика лікування пацієнтів з уперше виявленою множинною мієломою. А – кандидати для аутологічної трансплантації стовбурових клітин. Б – не кандидати для аутологічної трансплантації стовбурових клітин
Рис. 1. Тактика лікування пацієнтів з уперше виявленою множинною мієломою. А – кандидати для аутологічної трансплантації стовбурових клітин. Б – не кандидати для аутологічної трансплантації стовбурових клітин
Наразі стандартом лікування вперше діагностованої мієломи у хворих, які можуть бути кандидатами на трансплантацію стовбурових клітин, є режим VRd. За даними B.G.M. Durie та співавт. (2017), частота відповіді на проведене лікування, безрецидивна та загальна виживаність були значно кращими при використанні VRd у порівнянні з Rd [45]. Всі пацієнти, які отримують леналідомід, потребують медикаментозної профілактики тромбозу. Якщо режим VRd недоступний, можливе призначення VTd або VCd. Додатковою альтернативою для пацієнтів, які не є кандидатами на трансплантацію стовбурових клітин, є режим із використанням даратумумабу (DRd). Перспективною зарекомендувала себе комбінація Dara-VTd, проте остаточні результати впливу на загальну та безрецидивну виживаність наразі ще очікуються [48].
Основні опції для хворих, які не є кандидатами на трансплантацію стовбурових клітин, – це режими VRd та DRd. Застосування мелфалановмісних режимів не рекомендоване через ризик розвитку мієлодиспластичного синдрому та лейкозу.
Підтримувальна терапія показана після аутологічної трансплантації стовбурових клітин. Зазвичай з цією метою використовується леналідомід, що дозволяє істотно покращити безрецидивну та загальну виживаність, проте підвищує в 2-3 рази ризик розвитку злоякісних новоутворень інших локалізацій [47-53]. Іншою опцією є бортезоміб, вплив якого на загальну виживаність особливо виражений у пацієнтів із del(17p) [50].
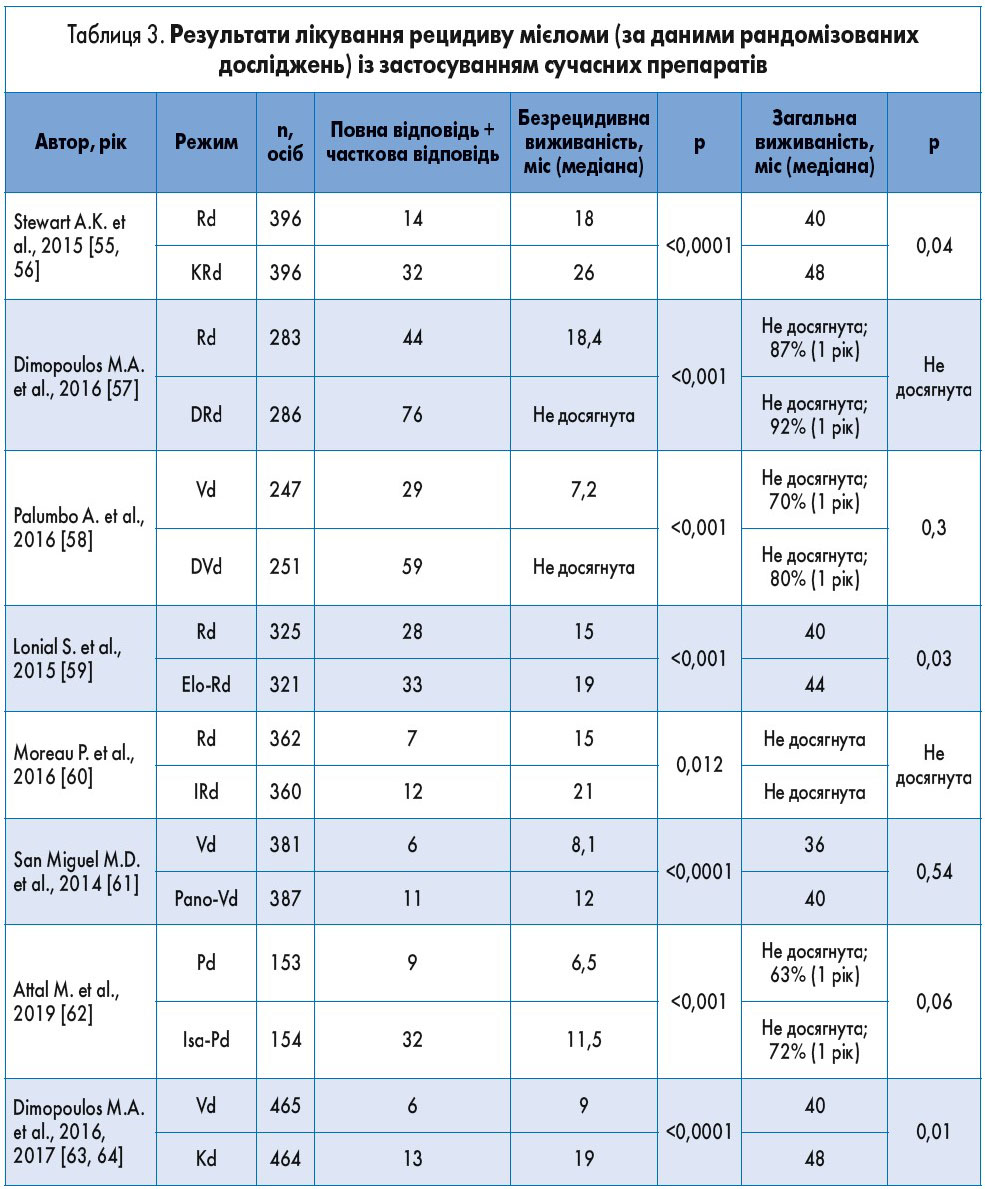
Практично у всіх пацієнтів із множинною мієломою з часом виникає рецидив. До впровадження даратумумабу медіана безрецидивної та загальної виживаності при рефрактерності до леналідоміду та бортезомібу була низькою [54]. Загальні принципи вибору тактики лікування наведені на рисунку 2, а результати ключових досліджень зведені в таблиці 3. Впровадження новітніх препаратів (у тому числі даратумумабу, карфілзомібу, ізатуксимабу, елотузумабу, панобіностату) дозволило істотно покращити як показники загальної відповіді на терапію, так і подовжити безрецидивну виживаність хворих з рецидивом мієломи.
 Рис. 2. Тактика лікування пацієнтів із рецидивом множинної мієломи. А – перший рецидив. Б – другий та наступні рецидиви
Рис. 2. Тактика лікування пацієнтів із рецидивом множинної мієломи. А – перший рецидив. Б – другий та наступні рецидиви
Висновки. Підсумовуючи вищезазначене, слід відмітити, що впровадження сучасних підходів до діагностики та стратифікації на групи ризику та лікування множинної мієломи (не лише первинної, а й рецидиву захворювання) дозволило значно поліпшити виживаність пацієнтів. Аутологічна трансплантація стовбурових клітин має застосовуватись у відповідної категорії хворих після індукційної терапії. Тривають дослідження, які потенційно можуть розширити показання до застосування новітніх лікарських засобів, у тому числі таргетної терапії.
Література
- Rajkumar S.V., Dimopoulos M.A., Palumbo A. et al. International Myeloma Working Group Updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014; 15: e538-e548.
- Roodman G.D. Pathogenesis of myeloma bone disease. Leukemia. 2009; 23: 435-441.
- Hillengass J., Usmani S., Rajkumar S.V. et al. International myeloma working group consensus recommendations on imaging in monoclonal plasma cell disorders. Lancet Oncol. 2019; 20: e302-e312.
- Short K.D., Rajkumar S.V., Larson D. et al. Incidence of extramedullary disease in patients with multiple myeloma in the era of novel therapy, and the activity of pomalidomide on extramedullary myeloma. Leukemia. 2011; 25: 906-908.
- Kyle R.A., Gertz M.A., Witzig T.E. et al. Review of 1,027 patients with newly diagnosed multiple myeloma. Mayo Clinic Proc. 2003; 78: 21-33.
…
64. Dimopoulos M.A., Goldschmidt H., Niesvizky R. et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017; 18: 1327-1337.
Підготувала Олена Поступаленко
CP-163394
Тематичний номер «Онкологія, Гематологія, Хіміотерапія» № 3 (64) 2020 р.