


20 жовтня, 2015
Малі імунодефіцитні хвороби: визначення, класифікація, клінічні прояви, діагностика і лікування (II частина)
Продовження. Початок у № 17.
Епідеміологія
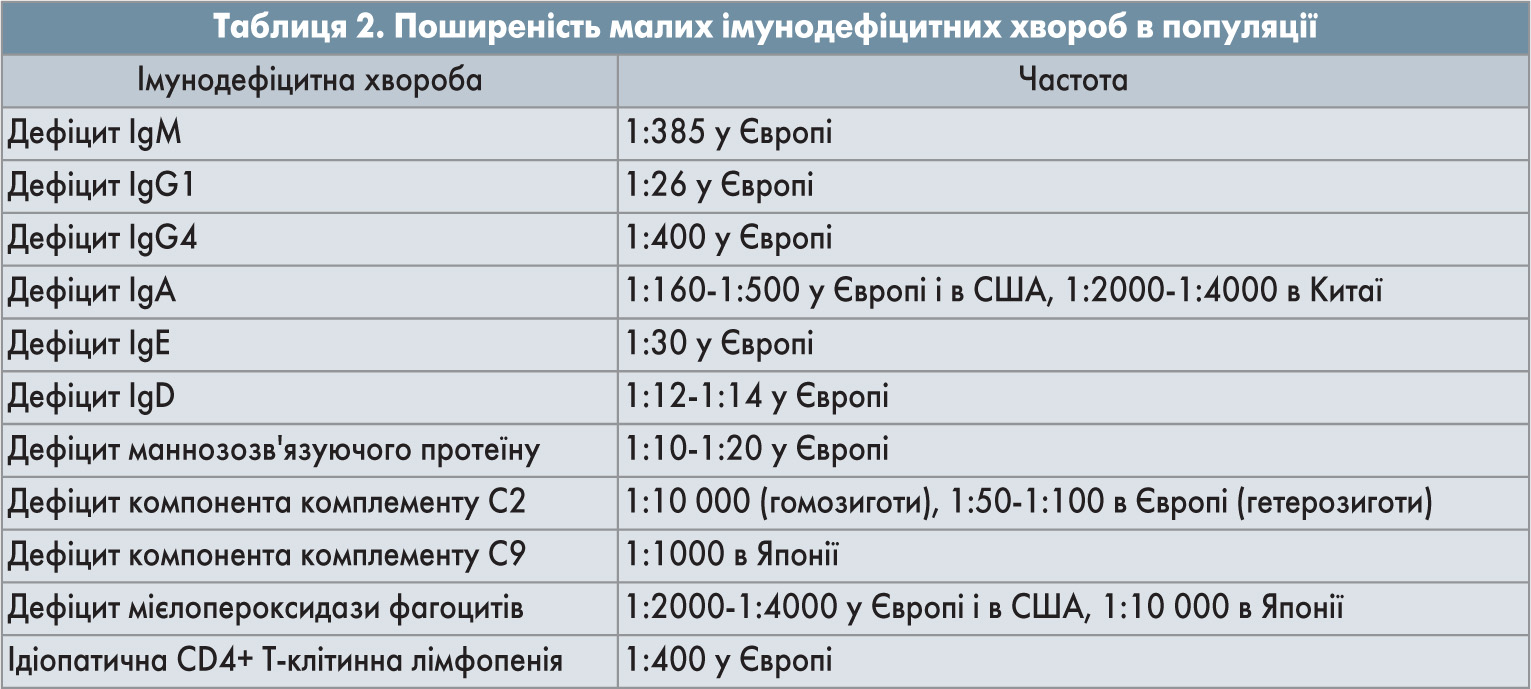
Малі імунодефіцитні хвороби є досить поширеними в людській популяції (табл. 2). Незважаючи на певні винятки, можна визначити закономірність: чим менша імунна недостатність лежить в основі імунодефіциту, тим більшою є поширеність цієї імунної дисфункції серед людей, що пов'язано з обмеженням дії природного відбору. Не виключено, що з цієї ж причини покращення якості життя і надання медичної допомоги парадоксально сприяють накопиченню малих імунодефіцитних хвороб у сучасній людській популяції. Наприклад, ізольований дефіцит IgD згідно з актуальними даними не зумовлює розвиток інфекційного синдрому і зрідка є причиною деяких аутоімунних та імунозапальних розладів у людей, включно з аутоімунним гепатитом і системним червоним вовчаком. Поширеність цього імунодефіциту дуже висока – на рівні 6-8% загальної популяції. Саме в безсимптомності перебігу вбачають причину обмеження дії природного відбору і феномена накопичення цього імунодефіциту серед людей. Проте існують чинники, що втручаються в розподіл імунодефіцитної хвороби в популяції. Йдеться про ефекти засновника і селективної переваги.
Ефект засновника реалізується в обмежених, закритих популяціях, де вперше з'явилася мутація, яка лежить в основі імунодефіцитної хвороби. Цей ефект, з одного боку, може сприяти накопиченню хвороби в певному регіоні через малу кількість схрещувань з представниками інших популяцій, а з іншого – обмежувати надходження генетичних хвороб з інших етносів. Класичним прикладом впливу ефекту засновника є японська популяція. Так, поширеність дефіциту мієлопероксидази фагоцитів у Західній Європі і США становить 1 випадок на 2-4 тис. мешканців, натомість у Японії – 1 випадок на 10 тис. людей. При цьому в західному світі хвороба зумовлена мутацією R569W, а в Японії – іншим генетичним порушенням. Ефект засновника також може зумовлювати неоднорідність поширення хвороби в одній географічній зоні у зв'язку з наявністю на її території субзон, де практикуються близькоспоріднені шлюби. Так, загалом дефіцит мієлопероксидази фагоцитів в Італії зустрічається в одного з 2 тис. мешканців, однак у провінції Брешія та в регіоні Фріулі-Венеція-Джулія частота хвороби досягає 1 випадку на 500 мешканців.
Ефект селективної переваги послаблює дію природного відбору у зв'язку з на перший погляд парадоксальною появою певних сприятливих, корисних властивостей, пов'язаних з імунодефіцитом. Навіть Х-зчеплена агаммаглобулінемія, важка форма первинного гуморального імунодефіциту, супроводжується природною резистентністю до вірусу Епштейна-Барр. При малих імунних дисфункціях ефект селективної переваги є більш вираженим. Так, накопичення дефіциту компонента комплементу С2 серед японців пов'язане, окрім інших причин, із захистом від розвитку інфекційно-токсичного шоку під час менінгококцемії. Дефіцит маннозозв'язуючого білка зустрічається майже з тією частотою, що й дефіцит IgD, однак часто призводить до розвитку клінічних симптомів, нерідко таких, які становлять загрозу для життя. Якщо малосимптомність є основною відомою причиною поширення ізольованого дефіциту IgD, то при дефіциті маннозозв'язуючого білка з урахуванням великої кількості клінічно маніфестних форм, здається, більшого значення набувають певні переваги, що їх надає ця імунна дисфункція, – наприклад, захист від пульмонального туберкульозу або обмеження вогнища ішемічного інсульту при цереброваскулярній хворобі.
У поширеності малих імунодефіцитних хвороб серед людей, як правило, не відзначається певних гендерних відмінностей, оскільки ці імунні дисфункції зрідка передаються за Х-зчепленим типом. Однак у деяких дослідженнях такі відмінності продемонстровані, що може пояснюватися впливом статевих гормонів. Так, показано, що застосування пероральних контрацептивів призводить до вторинного дефіциту природних кілерів у жінок, тому феномен впливу статевих гормонів теоретично може мати значення при маніфестації первинного дефіциту природних кілерів у представників різної статі. L.A. Hanson і співавт. встановили, що серед дітей на дефіцит субкласів IgG частіше страждають хлопчики (3 хлопчики / 1 дівчинка), натомість серед дорослих імунодефіцит здебільшого реєструється в жінок (1 чоловік / 3 жінки). D.S. Ahmad і співавт. повідомили про співвідношення чоловіків і жінок на рівні 1,8:1,0 серед пацієнтів з ідіопатичною СD4+ Т-клітинною лімфопенією.
Расові відмінності в частоті характерні не для всіх малих імунних дисфункцій. C. Feldman і співавт. у спеціально спланованому дослідженні не виявили суттєвих відмінностей у частоті та клінічних проявах ізольованого дефіциту субкласів IgG серед представників европеоїдної і негроїдної рас. Натомість вибірковий дефіцит IgА зустрічається з частотою 1:160-1:500 у європеоїдів і лише 1:2000-1:4000 серед монголоїдів.
Деякі імунні дисфункції більше проявляються в дитячому віці: наприклад, дефіцит IgA часто зустрічається серед дітей, однак до 10-12-річного віку щонайменше в половині випадків зазнає спонтанної компенсації. Ці так звані вікові імунодефіцити не мають бути підставою для невиправданої самозаспокоєності клініцистів, оскільки до моменту компенсації імунна дисфункція може призвести до розвитку важких ускладнень, наприклад бронхіальної астми або ювенільного ревматоїдного артриту. Деякі малі імунодефіцити, наприклад ідіопатична CD4+ Т-клітинна лімфопенія, частіше зустрічаються серед дорослих, ніж у дітей.
Клінічний дебют імунодефіциту можливий у будь-кому віці. Так, описано випадок розвитку потенційно летальних інтрацеребральних бактеріальних абсцесів у новонародженого з дефіцитом IgA, що вказує на безпрецедентно ранню маніфестацію хвороби; натомість М. Endoh і співавт. повідомили про перші прояви дефіциту IgМ у 85-річного пацієнта, якого раніше вважали імунокомпетентним. Крім того, імунодефіцити можуть видозмінювати клінічні прояви протягом онтогенезу: якщо у дітей переважають інфекційні симптоми, то в дорослих більшою є питома вага алергічних, аутоімунних і неопластичних ускладнень.
Спосіб життя, індивідуальний досвід взаємодії з мікроорганізмами та вплив додаткових несприятливих чинників суттєво модифікують важкість перебігу малих імунодефіцитних хвороб. Однак до оцінки потенційно шкідливих чинників слід підходити диференційовано при кожному імунодефіциті з урахуванням особливостей патогенезу хвороби. Так, куріння може поглиблювати наявний дефіцит субкласів IgG, як це показали в спеціальному дослідженні V. Popa і співавт. Натомість при дефіциті мієлопероксидази відзначається парадоксально знижений ризик формування раку легень унаслідок куріння, оскільки зазначений фермент бере участь в оксидації бензпірену з формуванням метаболітів-канцерогенів. Якщо іонізуюча радіація є важливим чинником формування вторинного дефіциту природних кілерів, що може зумовити поглиблення існуючого первинного імунодефіциту, то природні кілерні Т-клітини виявляють резистентність до променевої терапії.
Патогенез
Рецидивні інфекційні ураження є прямим наслідком дефіциту певного імунного чинника в імуноскомпрометованих пацієнтів, а їх частота і важкість визначаються вагомістю втраченого фактора в здійсненні захисної імунної відповіді проти мікроорганізмів, що атакують організм людини. Так, дефіцит маннозозв'язуючого білка призводить до послаблення розпізнавання маннозовмісних патогенів, зниження інтенсивності запалення, недостатності реакцій комплемент-опосередкованої цитотоксичності, що зумовлює розвиток інфекційних уражень, насамперед бактеріальних інфекцій. При дефіциті природних кілерів унеможливлюється реалізація реакцій спонтанної клітинно-опосередкованої цитотоксичності, що створює передумови для розвитку вірусних інфекцій. Хоча при гуморальних імунодефіцитах частіше розвиваються бактеріальні інфекції, а при клітинних – вірусні, ці відмінності не є абсолютними. Якщо IgG2 і IgG4 спрямовані здебільшого до полісахаридних антигенів, то IgG1 і IgG3 розпізнають переважно білкові молекули, в тому числі антигени вірусів. Тому К. Kallio-Laine і співавт. у спеціально спланованому дослідженні продемонстрували, що сироваткова концентрація IgG1 була нижчою (p=0,009), а частота виявлення ізольованого дефіциту IgG1 – більшою (p<0,001) у пацієнтів з рецидивним серозним менінгітом Молларе, викликаним вірусом простого герпесу 2 типу, у порівнянні з відповідними показниками в учасників контрольної групи. Ризик нового епізоду менінгіту зростав зі зниженням концентрації IgG1 (показник інциденту 2,05). Експресія алелей HLA-DRB1*01 і -B*27 була асоційована з дефіцитом IgG1 і високим ризиком нейроінфекції.
Розвиток алергічних, імунозапальних та аутоімунних ускладнень зумовлений порушенням реалізації імунних реакцій з підтримання толерантності до деяких ендогенних й екзогенних антигенів при дефіциті певних чинників імунітету, задіяних у реалізації механізмів імунної толерантності. Можна сказати, що в таких випадках відзначається реалізація шкідливої несанкціонованої імунної реакції через нездатність здійснити контролюючу імунну реакцію з пригнічення алергії й аутоімунітету. Для розвитку імунозалежних ускладнень необхідна наявність імунної недостатності такої глибини, що зумовить порушення реакцій з підтримання толерантності, однак збереже здатність імунної системи реалізувати імуноопосередковане аутоушкодження. Так, при тяжких комбінованих імунодефіцитах аутоімунні розлади майже не реєструються через драматичність інфекційних уражень та глибоку лімфопенію або навіть алімфоцитоз, пов'язані з нестачею аутореактивних лімфоцитів, здатних опосередкувати аутоімунітет. Натомість при малих імунних дисфункціях з неглибокою імунною недостатністю і в разі значною мірою редукованого інфекційного синдрому формуються сприятливі умови для розвитку імунозалежних ускладнень, важкість яких у багатьох випадках перевищує важкість власне інфекційних уражень.
При імунодефіцитних хворобах відзначаються різні механізми розвитку імунозалежних ускладнень. При одному й тому ж імунодефіциті і в одного й того ж пацієнта можуть реалізовуватися відмінні механізми формування алергії та аутоімунітету, тоді як при різних імунодефіцитах виникають імунозалежні ускладнення зі спільним механізмом розвитку. При гуморальних імунодефіцитах формуються переважно аутоімунні ускладнення з цитотоксичним та імунокомплексним механізмом розвитку, натомість при клітинних імунодефіцитах розвиваються здебільшого (але не виключно) клітинні реакції аутоімунітету. При фагоцитарних імунодефіцитах питома вага алергічних та аутоімунних ускладнень менша, ніж при порушеннях адаптивного імунітету.
Так, атопічні реакції, зумовлені компенсаторною гіперпродукцією IgE, типові для первинних дефіцитів інших класів/субкласів імуноглобулінів. При дефіциті IgА та IgG4 ризик розвитку атопічних реакцій підвищений також завдяки втраті блокуючої активності цих імуноглобулінів щодо IgE. При дефіциті природних кілерів і природних кілерних Т-лімфоцитів схильність до атопії пояснюють зниженою продукцією гамма-інтерферону, який зазвичай пригнічує імунні реакції, опосередковані Т-хелперами 2 типу. Сироваткова хвороба з імунокомплексним механізмом розвитку у відповідь на препарати крові та їх похідні при тотальному дефіциті IgА пов'язана з наявністю анти-IgА-антитіл в організмі таких пацієнтів. Ці імуноглобуліни зустрічаються принаймні в третині випадків тотального дефіциту IgА і не відзначаються в пацієнтів із парціальною формою імунодефіциту. Дефіцит природних кілерних Т-клітин зумовлює розвиток контактного алергічного дерматиту завдяки дефіциту інтерлейкіну-10 – протизапального цитокіну, активними продуцентами якого є зазначені лімфоцити. Цей цитокін пригнічує клітинну опосередковану Т-хелперами 1 типу імунну реакцію, що лежить в основі контактної гіперчутливості. Натомість при ідіопатичній CD4+ Т-клітинній лімфопенії частота контактного алергічного дерматиту знижена через послаблення реакцій сповільненої гіперчутливості в умовах дефіциту Т-хелперів 1 типу. Однак в разі ідіопатичної Т-лімфоцитопенії іноді більш вагому роль має дефіцит регуляторних Т-лімфоцитів з фенотипом CD4+CD25+, ніж дефіцит Т-хелперів 1 типу, що може пояснити, парадоксальні на перший погляд випадки розвитку контактного дерматиту при зазначеній імунній дисфункції. При дефіциті IgЕ відзначаються псевдоалергічні (псевдоатопічні) реакції, пов'язані з втратою стабілізуючого впливу IgЕ на мастоцити, що зумовлює підвищену чутливість останніх до неспецифічних стимулів, включно з медикаментами, механічними подразниками і біологічно активними речовинами.
Розвиток аутоімунних реакцій за цитотоксичним типом, включно з аутоімунними цитопеніями, при гіпоімуноглобулінемії, дефіциті тотального IgG та субкласів IgG можна пояснити порушенням функціонування сітки антиідіотипових антитіл, які нейтралізують аутоантитіла до моменту їх взаємодії зі специфічними аутоантигенами та модулюють активність аутореактивних В-лімфоцитів через вплив на їхні Fc-рецептори. Натомість імунокомплексні реакції, зокрема системний червоний вовчак та гломерулонефрит, при зазначених імунних дисфункціях пояснюються порушенням процесів солюбілізації та ресолюбілізації циркулюючих імунних комплексів, що послаблює кліренс останніх макрофагами печінки і селезінки. При дефіциті мукозального імунітету механізм розвитку аутоімунних ускладнень вбачають у масованому надходженні антигенів, в тому числі суперантигенів, через слизові оболонки, зокрема, внаслідок втрати нейтралізуючої активності IgА та послаблення захисної ексудативної реакції при дефіциті IgЕ. При дефіциті ініціальних білків системи комплементу імунокомплексні реакції зумовлені послабленим кліренсом імунних комплексів макрофагами, які розпізнають ці комплекси через рецептори до фрагментів зазначених білків комплементу. Натомість при дефіциті термінальних компонентів системи комплементу відзначається більша питома вага аутоімунних уражень з цитотоксичним і клітинним механізмом розвитку, включно з анкілозуючим спондилоартритом і ревматоїдним артритом, оскільки за фрагментами цих протеїнів здійснюється вилучення депозитів комплементу, що формують мембранатакуючі комплекси на мембранах клітин органів-мішеней. Маннозозв'язуючий лектин важливий у кліренсі циркулюючих суперантигенів та імунних комплексів, а також у видаленні апоптотичних клітин і тканинного детриту, що визначає розвиток системного червоного вовчаку переважно в пацієнтів з дефіцитом такої молекули. Імунорегуляторні порушення, пов'язані з недостатністю контактного механізму імунного відхилення та дефіцитом таких цитокінів, як інтерлейкін-10 та гамма-інтерферон, пояснюють аутоімунні ускладнення при дефіцитах природних кілерів та природних кілерних Т-клітин. У разі ідіопатичної CD4+ Т-клітинної лімфопенії причину аутоімунних уражень вбачають у дефіциті регуляторних Т-лімфоцитів, що в нормі підтримують анергію аутореактивних імунокомпетентних клітин в периферичних імунних органах.
Також алергічні та аутоімунні ураження в імуноскомпометованих пацієнтів можна пояснити аномальною персистенцією мікробних тригерів, що безпосередньо пов'язано з порушенням протиінфекційного імунітету. Так, інфекційно-залежна бронхіальна астма асоційована з частими епізодами вірусних респіраторних інфекцій, що призводить до гіперреактивності бронхіального дерева, а мультиформна еритема пов'язана з вірусом простого герпесу 1 типу. Розвиток DIHS/DRESS при дефіциті природних кілерів пояснюють втратою контролю над вірусами герпесу 6 і 7 типів у зоні резервації (слинні залози), а при гіпоімуноглобулінемії – сприятливими умовами для тривалого збереження стану вірусемії. Вірус Епштейна-Барр, який часто зазнає аномальної реактивації в імуноскомпрометованих пацієнтів, відомий як тригер низки аутоімунних хвороб, включно із системним червоним вовчаком, аутоімунним тиреоїдитом і розсіяним склерозом. Механізм розвитку аутоімунітету в такому разі вбачають у вірус-індукованій поліклональній активації В-лімфоцитів із залученням аутореактивних клонів. Інфекція, викликана Campylobacter jejuni, часто розвивається в пацієнтів з гуморальними імунодефіцитами, що визначає тісну асоціацію цих імунних дисфункцій із синдромом Гійєна-Барре і хронічною запальною демієлінізуючою полінейропатією, тригером яких є зазначений бактеріальний агент у зв'язку з феноменом молекулярної мімікрії. Персистенція бета-гемолітичних стрептококів групи А в ротоглотці, зумовлена сповільненим і/або незавершеним фагоцитозом, пояснює відому асоціацію дефіциту мієлопероксидази фагоцитів і хронічної ревматичної лихоманки. Натомість у розвитку хронічної інфекції, викликаної S. mutans, у зв'язку з послабленням опсонізації вбачають зв'язок дефіциту манозозв'язуючого протеїну і синдрому Бехчета в людській популяції.
Клінічні прояви
Перш за все, слід зазначити, що малі імунні дисфункції суттєво погіршують якість життя пацієнтів. G.H. Jоrgensen і співавт. у контрольованому дослідженні показали погіршення пов'язаної зі здоров'ям якості життя у пацієнтів з ізольованим дефіцитом IgA. Найбільш вагомими чинниками ризику низької якості життя були кількість курсів антибіотиків протягом року (p<0,001), кількість ліків, що приймаються щоденно (p<0,01), алергічний ринокон'юнктивіт (p<0,05), хронічні м'язово-скелетні симптоми (p<0,05) і тривога та/або безсоння (p<0,005).
Основою клінічного фенотипу імунодефіцитних хвороб є інфекційний, алергічний, імунозапальний, аутоімунний та неопластичний синдроми, однак можуть розвиватися і деякі додаткові прояви зі складним патогенезом, включно з психічними і кардіоваскулярними хворобами. Загалом можна виділити 3 основні синдроми: інфекційний, синдром порушення підтримання імунної толерантності (алергічні, імунозапальні, аутоімунні розлади) та неопластичний, що відповідає сучасним уявленням про основні функції імунної системи в організмі людини: здійснення протиінфекційного захисту, підтримання імунної толерантності та знищення пухлинних клітин. Не є виключенням і малі імунодефіцитні хвороби. Якщо в одного пацієнта розвиваються всі характерні синдроми протягом онтогенезу, то можна говорити про повний клінічний фенотип імунодефіциту. При малих імунодефіцитних хворобах частіше, ніж при класичних імунодефіцитах, формується частковий клінічний фенотип, коли розвиваються тільки деякі з перерахованих синдромів, хоча відомі й випадки розгорнутої клінічної картини.
Наразі необхідним є проведення великих епідеміологічних досліджень із залученням високоточних методів сучасної статистики для опису клінічного фенотипу різних малих імунодефіцитів і відокремлення від випадків механістичних поєднань. У цьому напрямі вже зроблені перші вагомі кроки. У нещодавньому великому епідеміологічному дослідженні Е. Magen і співавт. за участю 18 487 осіб показаний тісний зв'язок первинного дефіциту IgE з гіперреактивістю бронхіального дерева і бронхіальною астмою в дітей та бактеріальним середнім отитом, хронічним синуситом, аутоімунними синдромами і неоплазіями в дітей і дорослих. С.Н. Jorgensen і співавт. у контрольованому дослідженні за типом «випадок-контроль» продемонстрували, що пацієнти з дефіцитом IgA частіше за здорових осіб страждають на рецидивні інфекції верхніх і нижніх дихальних шляхів, причому у 25% випадків відзначається більше 1 випадку пневмонії протягом 2 років, натомість у загальній популяції подібне трапляється лише в 1,6% випадків (p<0,001). Алергічні та аутоімунні прояви мали місце у 84,4% випадків, а в контрольній групі – лише в 47,6% випадків (p<0,001).
Оскільки важкість імунодефіциту тісно корелює лише з інфекційним синдромом, усі інші прояви імунодефіцитної хвороби ми рекомендуємо визначати як імунозалежні ускладнення, адже їх розвиток визначається дією додаткових чинників, а замісна імунотерапія не завжди сприяє компенсації. Можна виділити закономірність: чим глибшою є імунна недостатність, тим більша питома вага в структурі клінічних проявів імунодефіциту належить інфекційним проявам і менша – імунозалежним ускладненням. Так, при Х-зчепленій агаммаглобулінемії алергічні й аутоімунні ускладнення зустрічаються не частіше, ніж у 30% випадків, при дефіциті IgA питома вага таких проявів може досягати 40-50%, тоді як при дефіциті IgE – 50-70%, а дефіцит IgD маніфестує виключно у вигляді аутоімунних розладів. N. Riyaz і співавт. описали у 27-річного пацієнта з тотальним дефіцитом IgE розвиток цукрового діабету 1 типу, pyoderma gangrenosum, мікроспоричного коліту й ідіопатичного гіпереозинофільного синдрому без суттєвих проявів інфекційних уражень як на момент госпіталізації до стаціонару, так і протягом анамнезу життя.
Далі буде