


1 лютого, 2022
Регданвімаб – інноваційний засіб на основі моноклональних антитіл для лікування COVID-19, який пройшов авторизацію ЕМА. Коли препарат стане доступним в Україні?
Моноклональні антитіла (mAbs) почали застосовувати в медицині з 1970-х років переважно для лікування онкологічних захворювань. Останнім часом у зв’язку з розвитком біотехнологій сфера їх застосування значно розширилася та включає лікування низки інфекційних захворювань. Уточнення патогенетичних механізмів розвитку коронавірусної хвороби (COVID-19) і створення mAbs проти SARS-CoV-2 визначило нові молекулярні мішені та сформувало умови для персоніфікованого підходу до противірусної терапії. У цьому огляді ми розглянемо механізм дії mAbs, які дозволені для лікування COVID-19, і наведемо найновіші докази їхньої клінічної ефективності.
У патогенезі COVID‑19 виділяють дві фази. Перша фаза розвивається внаслідок прямої цитопатичної дії вірусу SARS-CoV‑2, який потрапляє до клітини шляхом зв’язування з рецептором ангіотензинперетворювального ферменту‑2 (АСЕ2) за допомогою рецепторзв’язувального домену (RBD) S1-субодиниці спайкового глікопротеїну, тоді як трансмембранна S2-субодиниця зумовлює злиття вірусної оболонки з клітинною мембраною. Друга фаза спричинена гіперімунною відповіддю організму та характеризується широким спектром системних клінічних проявів різного ступеня тяжкості, котрі чітко корелюють із віком і наявністю в пацієнта чинників ризику. Відповідно, ключову роль відіграє адекватна терапія, скерована на пригнічення розвитку неконтрольованого процесу, індукованого вірусемією, й асоційованого з нею каскаду імунопатологічних реакцій [1].
Методи й засоби терапії залежать від фази COVID‑19 і стану хворого, але єдина тактика ведення та лікування пацієнтів на сьогодні досі відсутня. Отже, лікування пацієнтів із COVID‑19 є складним і малопередбачуваним процесом; потрібно постійно враховувати індивідуальну клінічну ситуацію, спираючись на чинні рекомендації з високим ступенем достовірності. З одного боку, лікарі всього світу стикаються з проблемами інтерпретації й імплементації результатів численних клінічних випробувань, що проводяться та публікуються з такою швидкістю, яка раніше не траплялася. З іншого боку, інформаційна база наукових доказів постійно оновлюється, що дає лікарям унікальну можливість застосовувати в щоденній клінічній практиці найбільш ефективні та безпечні методи лікування коронавірусної інфекції.
Терапевтичні мішені COVID‑19: що нового?
Молекулярний механізм розвитку COVID‑19, визначений як «цитокіновий шторм», дав можливість спочатку виділити напрями терапевтичного втручання, на які можна було би впливати за допомогою вже наявних фармацевтичних препаратів.
У червні 2020 року R. A. Siemieniuk і співавт. опублікували результати метааналізу 206 рандомізованих контрольованих досліджень (РКД) та 189 наукових публікацій, у яких оцінювалися 84 схеми лікування COVID‑19. Дані систематичного огляду показали, що кортикостероїди знижують смертність у разі тяжких форм COVID‑19; ремдесивір може зменшити потребу в штучній вентиляції легень (ШВЛ), але на смертність, найімовірніше, не впливає; інгібітори інтерлейкіну‑6 (тоцилізумаб і сарилумаб), а також інгібітори Янус-кіназ (барцитиніб і руксолітиніб) можуть зменшувати тривалість госпіталізації та час перебування на ШВЛ. Інші втручання (азитроміцин, лопінавір/ритонавір, протималярійні, протипаразитарні препарати та β-інтерферон) не мають остаточних доказів переваги застосування [2].
Станом на вересень 2021 року у світі було зареєстровано понад 2900 РКД, в яких активно вивчаються різні схеми терапії переважно тяжких та ускладнених форм COVID‑19 у госпіталізованих пацієнтів. І тільки близько 12% усіх зареєстрованих РКД оцінюють ефективність і безпеку застосування біологічної терапії – реконвалесцентної плазми, внутрішньовенних імуноглобулінів, рекомбінантних противірусних антитіл – попри сприятливий профіль безпеки й успішний історичний досвід застосування цих препаратів під час інших пандемій [3].
Зв’язок між вірусним навантаженням і тяжкістю захворювання в масштабних РКД також не вивчався, хоча вже було встановлено кореляцію між цими двома чинниками та відомо, що S‑глікопротеїн є головною мішенню для природних нейтралізувальних антитіл проти SARS-CoV‑2, котрі відіграють головну роль у формуванні адаптивного гуморального імунітету при COVID‑19. Подальше уточнення молекулярних механізмів патогенезу COVID‑19 і доклінічні дослідження на тваринних моделях дали змогу визначити специфічні для SARS-CoV‑2 вхідні рецептори як потенційні терапевтичні мішені. Зокрема, було виявлено, що одним із найперспективніших напрямів терапії є застосування mAbs, які блокують зв’язування SARS-CoV‑2 з рецептором ACE2, перешкоджають потраплянню вірусу до клітини-мішені та зупиняють його реплікацію (рис. 1) [4-7].
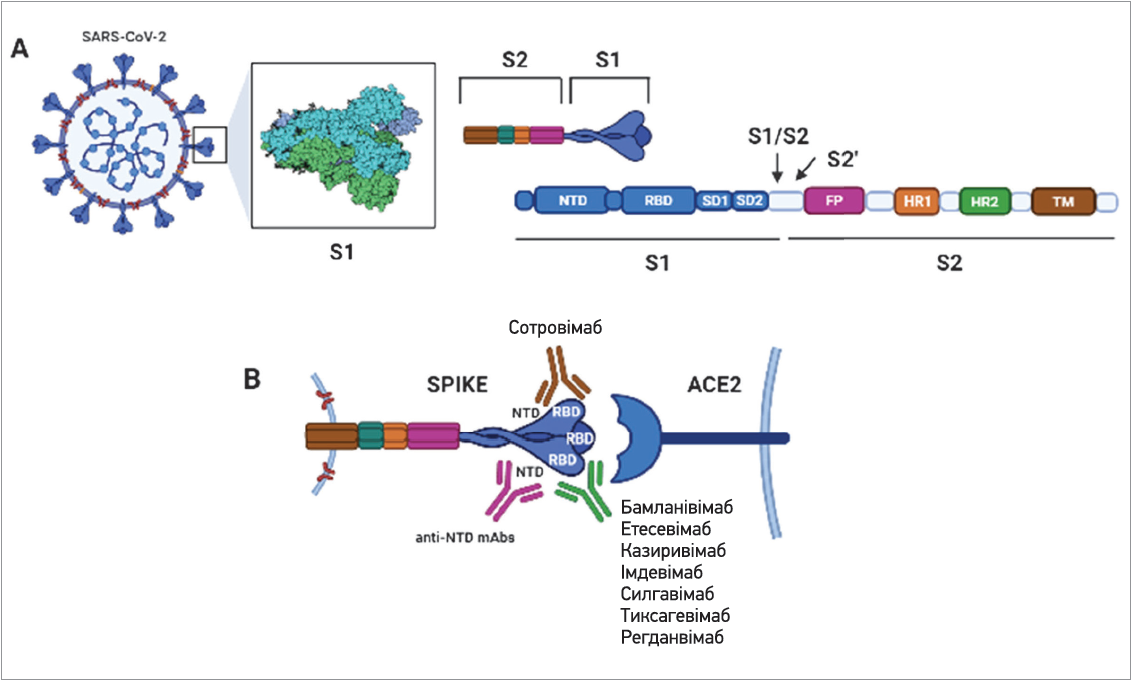
Рис. 1. Схематичне зображення S-протеїну SARS-CoV-2 та його взаємодій із клітинним рецептором (АСЕ2) і терапевтичними mAbs
Моноклональні антитіла проти SARS-CoV‑2
Загалом рекомбінантні mAbs є новим класом високоспецифічних біологічних препаратів, які успішно використовують для діагностики та лікування онкологічних, автоімунних, генетичних і деяких тяжких інфекційних захворювань: гарячки Ебола, сказу, гепатиту С, респіраторно-синцитіальної інфекції в дітей [5, 6].
Американське товариство з інфекційних хвороб (IDSA) розглядає застосування та вбачає велику перспективу mAbs проти RBD спайкового глікопротеїну SARS-CoV‑2 як терапевтичного та профілактичного засобу для амбулаторних пацієнтів із легкою та помірною COVID‑19, котрі належать до групи ризику ускладненого перебігу. Оновлені рекомендації IDSA ґрунтуються на таких нещодавно отриманих даних: швидке зменшення вірусного навантаження в організмі; специфічність і прогнозованість нейтралізувальної активності mAbs; можливість формування адекватної імунної відповіді швидше, ніж у разі вакцинації, коли на це потрібно декілька тижнів [8].
На відміну від препаратів реконвалесцентної плазми та внутрішньовенних імуноглобулінів, виробництво яких обмежене кількістю донорів, рекомбінантні mAbs не мають обмежень для масштабування після ідентифікації. За таких умов отримання доказів ефективності та безпеки застосування mAbs проти SARS-CoV‑2 і швидке розроблення практичних рекомендацій щодо зниження ризику тяжкого перебігу, госпіталізацій і смертності від COVID‑19 мають вирішальне значення для клініцистів. Відповідно, Управління з контролю якості продуктів харчування та лікарських засобів США (FDA), Європейське агентство з лікарських засобів (EMA) за процедурою EUA (Emergency Use Authorization) та інші національні регулятори з огляду на отримані докази схвалюють препарати mAbs проти SARS-CoV‑2 для лікування амбулаторних хворих на COVID‑19 із високим ризиком ускладненого перебігу.
Препарати mAbs для лікування COVID‑19: яка між ними відмінність
Казиривімаб/імдевімаб і бамланівімаб/етесевімаб – рекомбінантні нейтралізувальні mAbs імуноглобуліну людини (IgG1) з активністю проти SARS-CoV‑2. Обидва компоненти комбінацій mAbs зв’язуються з різними (які перекривають один одного) епітопами RBD S‑протеїну та блокують проникнення вірусу до клітин організму.
Комбінацію казиривімаб/імдевімаб дозволено до застосування в США, Європі, Великій Британії; препарат рекомендовано для лікування та профілактики COVID‑19 у дітей віком від 12 років (із масою тіла ≥40 кг) і дорослих. Всесвітня організація охорони здоров’я (ВООЗ) рекомендує використовувати комбінацію казиривімаб/імдевімаб у пацієнтів із легким перебігом COVID‑19 і високим ризиком госпіталізації, а також у серонегативних пацієнтів із тяжким перебігом. У таких випадках терапія проводиться разом з іншими препаратами в схемі лікування [9-12].
Комбінацію бамланівімаб/етесевімаб спочатку було схвалено в США для лікування та постконтактної профілактики COVID‑19 у дітей (у т. ч. новонароджених) і дорослих із високим ризиком тяжкого перебігу та/або госпіталізації. Надалі FDA переглянуло дозвіл і випустило інформаційний бюлетень, у якому рекомендує застосування комбінації бамланівімаб/етесевімаб тільки для постконтактної профілактики в амбулаторних пацієнтів. Своєю чергою, EMA спочатку повідомило, що препарат можна застосовувати пацієнтам від 12 років, які не потребують кисневої терапії, але мають високий ризик обтяження перебігу COVID‑19. Нині ЕMA зазначає, що пацієнти можуть отримувати комбінацію бамланівімаб/етесевімаб відповідно до національних рекомендацій [13, 14].
Сотровімаб – рекомбінантне mAb з активністю проти SARS-CoV‑2, яка забезпечується шляхом прикріплення до RBD S‑глікопротеїну вірусу та перешкоджанню його проникненню до клітини-мішені. Препарат схвалено у Великій Британії та США для лікування амбулаторних пацієнтів віком понад 12 років (із масою тіла ≥40 кг) із COVID‑19, що мають високий ризик прогресування хвороби та/або госпіталізації. Відповідно до критеріїв екстреного використання ЕМА схвалило застосування сотровімабу для лікування пацієнтів від 12 років, які не потребують кисневої підтримки, але мають високий ризик прогресування COVID‑19 [15-17].
Регданвімаб – рекомбінантне mAb IgG1, що зв’язується з епітопом RBD S‑глікопротеїну SARS-CoV‑2 і перешкоджає взаємодії з клітинним рецептором АСЕ2. Блокада з’єднання рецепторів RBD й АСЕ2 запобігає проникненню вірусу крізь мембрану клітини-мішені та запобігає його реплікації. Регданвімаб одним із перших mAbs отримав централізовану авторизацію ЕМА (жовтень 2020 року). Навесні 2021 року європейський Комітет із питань лікарських засобів для використання людиною (CHMP) EMA зробив висновок, що користь регданвімабу переважає ризики його застосування за схваленими показаннями. Прискорення авторизації в ЄС стало можливим завдяки тому, що CHMP уже розглянув дані щодо регданвімабу в ході циклічного огляду (rolling review). Препарат рекомендовано для лікування амбулаторних дорослих пацієнтів із COVID‑19, які не потребують кисневої підтримки, але мають високий ризик прогресування й ускладнення інфекції. У таких пацієнтів раннє лікування регданвімабом дає змогу зменшити симптоми коронавірусної інфекції та запобігти госпіталізації [18, 19].
У вересні 2021 року препарат було повністю схвалено в Республіці Корея для лікування COVID‑19 у пацієнтів віком понад 50 років із принаймні одним чинником ризику (ожиріння, хронічні серцево-судинні, легеневі, ниркові, печінкові хвороби, діабет і застосування імуносупресорів), а також у пацієнтів із помірними симптомами. Станом на листопад 2021 року курс терапії регданвімабом отримали 21 366 пацієнтів у 127 лікарнях Республіки Корея [20]. Дозвіл на клінічне застосування регданвімабу розглядають регуляторні органи США, Великої Британії, Канади й Австралії [21-23].
У систематичному огляді N. Kreuzberger і співавт. (2021) було проведено порівняльний аналіз плацебо-контрольованих РКД, у яких вивчалися ефективність і безпека mAbs (бамланівімаб/етесевімаб; казиривімаб/імдевімаб; сотровімаб; регданвімаб) проти SARS-CoV‑2, схвалених FDA й EMA для лікування пацієнтів різних віку, статі й етнічної приналежності з COVID‑19 легкого, середнього й тяжкого ступенів. Ефективність і безпеку застосування mAbs для специфічної терапії COVID‑19 оцінювали за такими критеріями: вплив на симптоми; госпіталізація; скорочення кількості смертей від усіх причин на 30-й і 60-й день; небажані ефекти (НЕ) та серйозні побічні явища (СПЯ). За цими критеріями визначили шість РКД за участю 17 495 пацієнтів; усі дослідження були завершені в період із липня по грудень 2021 року; цільова вибірка коливалася від 1020 до 10 000 учасників, середній вік яких становив 42-53 роки (амбулаторні пацієнти в чотирьох РКД) і 61 рік (госпіталізовані пацієнти у двох РКД).
Результати систематичного огляду продемонстрували, що в госпіталізованих пацієнтів (РКД ACTIVE‑3, RECOVERY) бамланівімаб може збільшувати ризик розвитку тяжкого перебігу COVID‑19 і виникнення НЕ через 5 днів після лікування та не впливає на час до виписки зі стаціонара. Комбінація казиривімаб/імдевімаб практично не впливає на потребу в ШВЛ і госпітальну смертність порівняно з іншими схемами лікування; даних щодо розвитку НЕ та СПЯ не виявлено.
Щодо амбулаторних пацієнтів (РКД BLAZE‑1, Weinreich, COMET-ICE, Eom‑21/NCT04602000), то всі досліджувані mAbs у режимі монотерапії знижують ризик госпіталізації та смертність протягом 30 днів після лікування (дані щодо смертності впродовж 60 днів у РКД не вивчалися), а також зумовлюють менше НЕ порівняно з плацебо. Застосування комбінації бамланівімаб/етесевімаб може спричинити більшу кількість серйозних НЕ порівняно з плацебо, не впливаючи при цьому на симптоми COVID‑19. Сотровімаб може зменшувати кількість пацієнтів, що потребують кисневої терапії; вплив на розвиток НЕ та СПЯ є незначним. Регданвімаб у будь-якій дозі (40 і 80 мг/кг) зменшує кількість госпіталізацій і смертність; доза 80 мг/кг має менший ризик розвитку НЕ (всі ступені), тоді як доза 40 мг/кг їх практично не спричиняє [24].
Регданвімаб: ключові дослідження клінічної ефективності та безпеки застосування
У середині 2021 року вийшов пресреліз щодо результатів фази багатонаціонального подвійного сліпого плацебо-контрольованого дослідження ІІ/ІІІ фази (NCT04602000), яке оцінювало ефективність і безпеку застосування таргетного mAb CT-P59 (регданвімаб) в амбулаторних пацієнтів із легким і середньої тяжкості перебігом COVID‑19 відповідно до стандартів терапії. У дослідженні взяли участь 1315 пацієнтів на базі 38 глобальних клінічних сайтів. У пацієнтів із легким і середньої тяжкості перебігом COVID‑19 і ризиком ускладнення застосування регданвімабу впродовж 7 днів від часу появи симптомів на 72% (р<0,0001) знижувало ризик госпіталізації або смерті до 28-го дня захворювання (первинна кінцева точка): 3,1% проти 11,1% у групі плацебо. Також у групі регданвімабу спостерігалася стійкіша редукція симптомів; час до одужання скоротився щонайменше на 4,2 дня; в пацієнтів із пневмонією прогресування хвороби знизилося на 50% порівняно з групою плацебо [25].
У ході першої частини дослідження (NCT04602000; EudraCT: 2020-003369-20) його учасники були рандомізовані на три групи: СТ-Р59 (регданвімаб) у дозах 40 і 80 мг/кг і плацебо; всі препарати отримувалися у вигляді 90-хвилинної внутрішньовенної інфузії. Вихідні демографічні та клінічні характеристики між групами були загалом зіставними: середній вік учасників – 51 рік; 87,5% – білі, 12,5% – азіати; жінки – 49,2%; середній ступінь тяжкості COVID‑19 мали 57,8% учасників.
Інфекцію SARS-CoV‑2 кількісно було підтверджено за допомогою полімеразної ланцюгової реакції (ПЛР). Клінічні прояви, що тривали 7 днів до введення досліджуваного препарату, включали гарячку, закладеність носа, кашель, задишку, біль у горлі та/або м’язах, голові, втому, втрату смаку та/або нюху, діарею тощо.
Результати першої частини РКД виявилися багатонадійними за всіма досліджуваними критеріями. Середній час до конверсії (95% довірчий інтервал – ДІ) у негативний результат ПЛР‑тесту до 28-го дня (первинна кінцева точка) становив 12,8 дня (9,00-12,84) у групі CT-P59 40 мг/кг; 11,9 дня (8,94-12,91) – у групі CT-P59 80 мг/кг; 12,9 дня (12,75-13,99) – у групі плацебо (рис. 2). Частка пацієнтів, які досягли конверсії в негативний результат ПЛР‑тесту, в групах регданвімабу (обидві дози) становила 92,1 та 87,4% порівняно з 83,5% у групі плацебо.
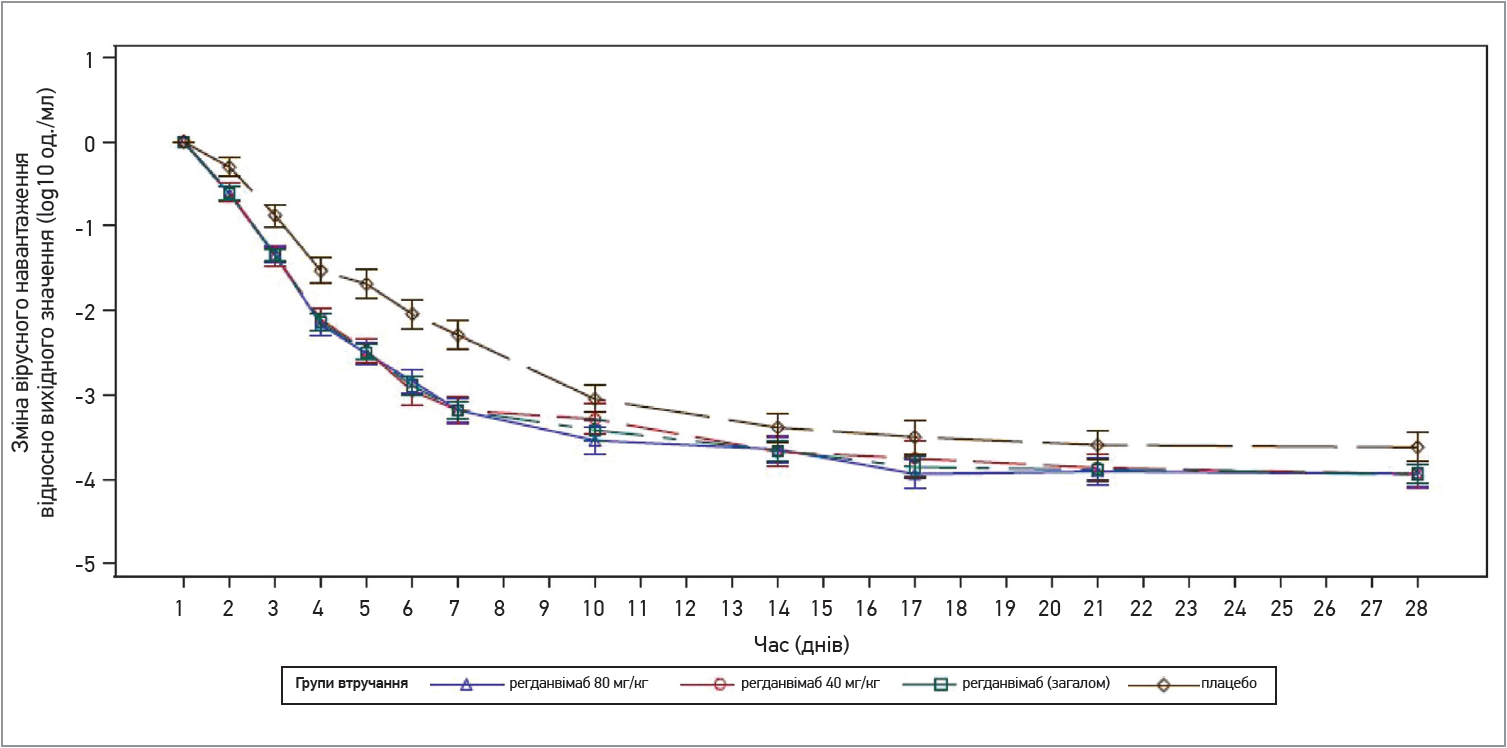
Рис. 2. Динаміка вірусного навантаження (кількість вірусних часточок) у мазках із носоглотки на тлі застосування регданвімабу та плацебо
Середній термін (95% ДІ) клінічного одужання (копервинна кінцева точка): 5,4 дня (3,97-6,78) у групі CT-P59 40 мг/кг; 6,2 дня (5,53-7,85) у групі CT-P59 80 мг/кг; 8,8 дня (6,72-11,73) у групі плацебо. Частка (95% ДІ) пацієнтів, які потребували госпіталізації або кисневої підтримки, виявилася меншою на 4% (2,1-10,9%) у групі СТ-P59 40 мг/кг і на 4,9% (2,1-10,9%) у групі СТ-P59 80 мг/кг порівняно з 8,7% (4,7-15,8%) у групі плацебо.
У ході дослідження регданвімаб продемонстрував сприятливий профіль безпеки; жодних серйозних НЕ, СПЯ та/або смертей в обидвох групах (40 і 80 мг/кг) не було. За результатами цього РКД дослідники дійшли висновку, що застосування mAb регданвімабу значно прискорює елімінацію SARS-CoV‑2 з організму та клінічне одужання, добре переноситься пацієнтами з COVID‑19 легкого та середнього ступенів тяжкості [26].
Раніше в подвійному сліпому плацебо-контрольованому дослідженні І фази (NCT04593641; n=18) регданвімаб у різних дозах (20, 40, 80 мг/кг) продемонстрував високу активність проти SARS-CoV‑2 у дорослих пацієнтів із легким перебігом COVID‑19. У контрольних мазках із носоглотки на 14-й день після застосування регданвімабу було виявлено більше зниження титрів вірусу порівняно з групою плацебо. У пацієнтів із високим початковим рівнем вірусного навантаження (105 копій/мл) застосування регданвімабу сприяло швидкій елімінації SARS-CoV‑2. На 14-й день у всіх пацієнтів (за винятком одного, що одержував плацебо) було констатовано клінічне одужання, середній час якого становив 3,39 дня в групі регданвімабу (всі дози) проти 5,25 дня в групі плацебо. У ході дослідження не було зареєстровано жодних реакцій, пов’язаних з інфузією, СПЯ або НЕ, що призвели до припинення лікування. Також надзвичайно важливим результатом стала відсутність феномену антитілозалежної цитотоксичності. Застосування регданвімабу не супроводжувалося жодними клінічно значущими порушеннями життєво важливих показників, гіперчутливістю, змінами на електрокардіограмі та під час інших інструментальних обстежень [27].
Ефективність і безпека застосування регданвімабу в щоденній клінічній практиці оцінювалася в ретроспективному когортному РКД. У дослідженні взяли участь понад 970 госпіталізованих пацієнтів віком ≥18 років із COVID‑19 легкої та середньої тяжкості й високим ризиком розвитку ускладнень. Чинниками високого ризику було визначено вік ≥60 років та/або наявність ≥1 основного хронічного захворювання: серцево-судинного (включно з гіпертензією), респіраторного (включно з астмою), діабет. Середня ступінь тяжкості COVID‑19 оцінювалася за наявністю пневмонії.
Результати дослідження продемонстрували, що терапія регданвімабом асоціювалася з істотним (р<0,001) зниженням частки пацієнтів з ускладненнями або смертю (первинна кінцева точка) порівняно з пацієнтами, котрі не отримували регданвімаб: 5,0% проти 21,5%. Також було виявлено, що краща відповідь на терапію регданвімабом спостерігалася в чоловіків похилого віку з високим індексом маси тіла та пневмонією [28].
Нині тривають постмаркетингові обсерваційні дослідження клінічної ефективності та безпеки регданвімабу, результати яких очікуються пізніше.
Висновки
За більш ніж 2 роки від часу початку пандемії COVID‑19 її так і не вдалося приборкати. У зв’язку з появою нових штамів SARS-CoV‑2 ефективність вакцинації суттєво знизилася. Наразі в більшості країн спостерігається бурхливе зростання захворюваності, спричинене новим штамом омікрон. Оскільки ефективних противірусних препаратів так і не було створено, одним із найперспективніших напрямів терапії стало застосування mAbs, що блокують зв’язування SARS-CoV‑2 з рецептором ACE2, перешкоджають потраплянню вірусу до клітини-мішені й зупиняють його реплікацію. Це зменшує вірусне навантаження та запобігає розвитку тяжких форм захворювання.
Одним із найвивченіших представників mAbs є регданвімаб, застосування котрого продемонструвало суттєве зниженням частки випадків тяжкого перебігу COVID‑19 і смертності порівняно з плацебо. Можливість застосовувати при пневмонії та широке терапевтичне вікно в 7днів від початку симптомів – це те, що на сьогоднішній день вирізняє регданвімаб з-поміж інших mAbs та може мати вирішальне значення в лікуванні пацієнтів із високим ризиком тяжкого перебігу COVID-19. Регданвімаб уже авторизований ЕМА, тож варто сподіватися, що й українські медики невдовзі поповнять свій арсенал засобів для лікування COVID-19.
Список літератури знаходиться в редакції.
Підготував Олег Сергійченко
