


15 травня, 2019
Болезнь Фабри: от диагноза к лечению
Болезнь Фабри (БФ) – наследственное заболевание, которое связано с нарушением метаболизма сфинголипидов и относится к группе лизосомных болезней накопления. Для БФ характерна высокая клиническая вариабельность, что значительно затрудняет своевременную диагностику и лечение. Опытом выявления и ведения лиц с БФ делились участники конференции «Наследственные орфанные заболевания – междисциплинарные диалоги», посвященной 10-летию Центра орфанных заболеваний НДСБ «ОХМАТДЕТ» МЗ Украины (1‑2 ноября 2018 года, г. Киев).
 О причинах, симптомах, возможных осложнениях и современных подходах к лечению БФ рассказала Сирен Сезер, профессор кафедры нефрологии Университета Баскент (Анкара, Турция).
О причинах, симптомах, возможных осложнениях и современных подходах к лечению БФ рассказала Сирен Сезер, профессор кафедры нефрологии Университета Баскент (Анкара, Турция).
Профессор Сезер напомнила слушателям, что БФ обусловлена полной или частичной недостаточностью лизосомной гидролазы – альфа-галактозидазы А (α-Gal A), что приводит к патологическому накоплению жирового вещества, известного как глоботриаозилцерамид (GL‑3). В норме α-Gal A отщепляет от GL‑3 остаток галактозы с образованием лактозилцерамида. Накопление GL‑3 происходит в различных типах клеток, что приводит к нарушению их функций, прогрессирующему поражению тканей и органов.
Частоту БФ оценивают в широком диапазоне – от 1/47600 до 1/117000 в общей популяции. Однако реальные показатели, очевидно, выше, поскольку атипичные формы заболевания часто не распознаются.
БФ сцеплена с Х-хромосомой – здесь расположен ген GLA, кодирующий α-Gal A. У мужчин, унаследовавших такую Х-хромосому, наблюдается классический фенотип БФ. Женщины, имеющие дефектную копию гена GLA в одной Х-хромосоме, обычно являются носителями заболевания, или течение болезни у них носит атипичный характер (при инактивации Х-хромосомы, несущей нормальный аллель GLA).
С. Сезер отметила, что на сегодняшний день известно около 1000 мутаций гена GLA. Их влияние на функциональную активность α-Gal A обусловлено, прежде всего, нарушением пространственной структуры фермента. Тяжесть проявлений заболевания зависит от остаточной активности дефектной α-Gal A: чем она ниже, тем раньше манифестирует патология, тем более выражены симптомы, тем значительнее поражены органы.
Соответственно, выделяют несколько типов мутаций гена GLA:
- патогенетические мутации, которые приводят к синтезу нефункционального α-Gal A и связаны с классическим фенотипом БФ;
- мутации, ассоциированные со снижением функциональной активности α-Gal A, которые служат причиной более поздней манифестации БФ;
- мутации неясного значения (VUS);
- полиморфизм гена, не влияющий на активность α-Gal A.
Одна и та же мутация может вызывать различную симптоматику БФ.
Накопление в клетках GL‑3 и его деацетилированной формы глоботриаозилсфингозина (lyso-GL‑3) ведет к нарушению функций клеток сосудистого эндотелия, гладкой мускулатуры сосудов, миокарда, фиброцитов клапанов сердца, эпителиальных клеток канальцев почек, нейронов и др. Повреждение тканей при БФ, вероятно, связано с иммунной реакцией на GL‑3 и lyso-GL‑3, которая обусловливает развитие хронического воспаления с последующим фиброзом и склерозом пораженных тканей (Rozenfel P., Feriozzi S., 2017).
Докладчик обратила внимание слушателей на симптомы БФ. При классическом фенотипе накопление GL‑3 происходит еще в пренатальный период, а первые проявления, такие как cornea verticillata, или воронковидная (вихревая) кератопатия, могут возникать сразу после рождения. В первые годы жизни также могут появляться нейропатическая боль, замедление роста, нарушения со стороны желудочно-кишечного тракта (ЖКТ), гипо- или ангидроз, кризы Фабри (атаки жгучей боли в ступнях и ладонях, распространяющиеся на другие части тела), непереносимость жары или холода, шум в ушах и головокружения, ангиокератомы (мелкие красновато-фиолетовые безболезненные папулы на коже). Как правило, до 16 лет также развиваются нарушения слуха, повышенная утомляемость, первые признаки ренальных и сердечно-сосудистых нарушений (Hopkin R.J. et al., 2016). У каждого ребенка с какими-либо из указанных симптомов следует исключить БФ.
До 30 лет обычно происходит дальнейшее прогрессирование симптомов, наблюдаются протеинурия, кардиомиопатия, транзиторные ишемические атаки. В возрасте после 30 лет состояние больных продолжает ухудшаться, резко повышается риск инсульта, сердечных заболеваний (тромбоз левого желудочка, стенокардия, аритмия). О неклассическом (атипичном) фенотипе БФ говорят при позднем начале заболевания, а также изолированных поражениях головного мозга, сердца или почек.
Профессор Сезер указала, что, согласно исследованию исходов при БФ (Fabry Outcome Survey [FOS]), заболевание может долго оставаться нераспознанным, что нередко заканчивается для пациента фатально (Mehta A. et al., 2004).
В связи с высокой клинической вариабельностью БФ, диагностика должна включать комплексную оценку клинической картины, лабораторные тесты и другие исследования. L. Van der Tol et al. (2015) для подтверждения диагноза БФ рекомендуют использовать следующие критерии:
- у мужчин: мутация в гене α-Gal A + классический фенотип, снижение активности α-Gal A в лейкоцитах <5% + один из симптомов (нейропатическая боль, cornea verticillata, ангиокератомы, повышение уровня GL‑3 и lyso-GL‑3 в плазме) или диагноз БФ у кого-либо из членов семьи;
- у женщин: мутация в гене α-Gal A + один из симптомов (нейропатическая боль, cornea verticillata, ангиокератомы, повышение уровня GL‑3 и lyso-GL‑3 в плазме) или диагноз БФ у кого-либо из членов семьи;
- стандартом диагностики БФ является измерение активности α-Gal A в сухих пятнах или лейкоцитах крови.
После установления диагноза БФ следует обязательно провести скрининг среди родственников больного – таким образом можно выявить в среднем 5 новых пациентов. По мнению докладчика, необходимо выполнять скрининг больных из групп высокого риска: находящихся на гемодиализе; с гипертрофической кардиомиопатией; после криптогенного инсульта.
Лечение пациентов с БФ должно быть направлено на предупреждение необратимого поражения тканей и органов. Для этого целесообразно проведение фермент-заместительной терапии (ФЗТ) и симптоматического лечения органной патологии. При ФЗТ используют рекомбинантные человеческие альфа-галактозидазы А, представленные в мире двумя препаратами: агалсидазой альфа и агалсидазой бета.
С. Сезер подчеркнула, что у лиц с классическим фенотипом БФ, диагностированным до 18 лет, ФЗТ обычно начинают в детстве (Hopkin R.J. et al., 2016). Согласно A. Ortiz et al. (2018), взрослым пациентам ФЗТ рекомендована:
- мужчинам, несущим мутации, связанные с классическим фенотипом БФ, независимо от наличия симптомов;
- женщинам, несущим мутации, ассоциированные с классическим фенотипом БФ, с симптомами поражения важных органов и систем (периферическая, центральная нервная система [ЦНС], сердце, почки, ЖКТ, кожа);
- женщинам, несущим мутации, связанные с классическим фенотипом БФ, без типичных симптомов, но с признаками поражения почек, сердца, ЦНС, выявляемыми лабораторно, гистологически или методами визуализирующей диагностики (снижение скорости клубочковой фильтрации [СКФ] <90 мл/мин/1,73 м2; альбуминурия >30 мг/г, «стирание» ножек подоцитов, гломерулосклероз; признаки «немого» инсульта при магнитно-резонансной томографии [МРТ] мозга, стенокардия, кардиофиброз и т.д.);
- мужчинам и женщинам, несущим мутации, связанные с более поздней манифестацией БФ, или мутации VUS, без типичных симптомов, но с признаками поражения почек, сердца, ЦНС, выявляемыми лабораторно, гистологически или методами визуализирующей диагностики (см. выше).
Опыт ФЗТ на протяжении 10 лет с применением агалсидазы бета (препарат Фабразим®*) по 1 мг/кг каждые 2 недели (К2Н) у пациентов с БФ показал, что наибольший эффект ФЗТ отмечался у лиц с незначительным поражением почек, у которых лечение было начато в более раннем возрасте. У больных, начавших ФЗТ в более позднем возрасте и/или страдавших тяжелым поражением почек, заболевание продолжало прогрессировать (Germain D.P. et al., 2015). В исследовании эффективности ФЗТ на протяжении 5 лет у детей с БФ было показано, что долгосрочная ФЗТ обеспечивает полное очищение от GL‑3 мезангиальных и эндотелиальных клеток почечных клубочков, а также подоцитов (Tondel C. et al., 2013). У пациентов с кардиомиопатией для долгосрочного улучшения морфологии и функциональных показателей миокарда ФЗТ следует начинать до развития фиброза миокарда (Weidemann F. et al., 2009).
Докладчик обратила внимание слушателей на исследование, в котором на протяжении года изучали последствия снижения дозы агалсидазы бета (до 0,3‑0,5 мг/кг) или переключения терапии на 0,2 мг/кг агалсидазы альфа при ФЗТ (Krämer J. et al., 2017). Если в контрольной группе пациентов, получавших обычную дозу агалсидазы бета, симптомы и состояние органов оставались стабильными, то в группе агалсидазы бета в пониженной дозе наблюдалось снижение СКФ, а в группе агалсидазы альфа – повышение медианного соотношения альбумин/креатинин. Кроме того, в группе снижения дозы и переключения отмечалось увеличение частоты болевых кризов и нарушений со стороны ЖКТ.
Таким образом, результаты исследований показали, что пациенты, получавшие стандартные дозы агалсидазы бета, имели относительно стабильный ход заболевания, но уменьшение дозы или переключение на агалсидазу альфа приводило к снижению уровня рСКФ и усилению симптомов, связанных с БФ. Впоследствии некоторые из этих пациентов вновь были переведены на агалсидазу бета (в полной дозе).
 Об особенностях лабораторной диагностики БФ рассказала сотрудник лаборатории медицинской генетики СМГЦ НДСБ «ОХМАТДЕТ», кандидат биологических наук Наталия Иосифовна Мыцик.
Об особенностях лабораторной диагностики БФ рассказала сотрудник лаборатории медицинской генетики СМГЦ НДСБ «ОХМАТДЕТ», кандидат биологических наук Наталия Иосифовна Мыцик.
Н.И. Мыцик отметила, что абсолютно специфичного теста на БФ не существует, поэтому диагностика БФ требует комплексного обследования с использованием всего доступного арсенала методов. Лабораторная диагностика БФ включает определение активности α-Gal A**, уровень накопления метаболитов GL‑3 и lyso-GL‑3, молекулярно-генетический анализ для выявления мутаций в гене GLA (Gal A. et al., 2011). У мужчин с классическим фенотипом БФ обычно наблюдается дефицит ферментативной активности (<10% от среднего референтного значения); с атипичным фенотипом при более легких формах – остаточная ферментативная активность, не превышающая нижнего значения референтного интервала. У женщин, независимо от фенотипа, активность α-Gal A может быть как пониженной, так и нормальной, поэтому данный показатель малоинформативен. У обоих полов активность α-Gal A не коррелирует с тяжестью клинических проявлений БФ.
Уровень GL‑3 в моче у мужчин с классическим фенотипом БФ повышен и является хорошим биомаркером, прежде всего, при нефрологической манифестации БФ. Однако при кардиальных формах БФ у мужчин и женщин, независимо от фенотипа, уровень GL‑3 в моче может находиться в пределах нормы. У большинства пациентов обоих полов с БФ информативен уровень lyso-GL‑3 в плазме (у мужчин в 5‑10 раз выше, чем у женщин), однако он не коррелирует с тяжестью клинических проявлений БФ. Показатели GL‑3 и lyso-GL‑3 не могут быть использованы для прогноза течения БФ, однако позволяют проводить индивидуальный мониторинг прогрессирования БФ или эффекта лечения.
Докладчик подытожила, что у мужчин лабораторная диагностика БФ должна начинаться с определения активности α-Gal A. При ее значениях >10% от среднего показателя референтного интервала проводят изучение мутаций в гене GLA. У женщин целесообразно сразу выполнять данный анализ.
 Заведующая Центром орфанных заболеваний НДСБ «ОХМАТДЕТ» МЗ Украины, кандидат медицинских наук Наталия Александровна Пичкур представила участникам конференции проект клинических рекомендаций по лечению БФ, разработанный ведущими отечественными специалистами.
Заведующая Центром орфанных заболеваний НДСБ «ОХМАТДЕТ» МЗ Украины, кандидат медицинских наук Наталия Александровна Пичкур представила участникам конференции проект клинических рекомендаций по лечению БФ, разработанный ведущими отечественными специалистами.
Н.А. Пичкур отметила, что Проект клинических рекомендаций по лечению БФ (далее – Проект) размещен на сайте Государственного экспертного центра МЗ Украины для общественного обсуждения (URL: http://mtd.dec.gov.ua/images/dodatki/KN/obg/2018_10_25_KN_Fabri.pdf).
Проект содержит:
- подробные характеристики клинических проявлений классической БФ с указанием типичного возраста появления;
- варианты БФ в зависимости от наличия фенотипических признаков (классический, неклассический, VUS, носительство);
- клинические особенности БФ в зависимости от пола;
- манифестацию БФ в разных возрастных группах (наиболее частые симптомы);
- критерии диагноза БФ.
Долгосрочное ведение взрослых пациентов с БФ должно включать своевременное назначение ФЗТ, регулярную оценку прогрессирования заболевания и применение надлежащих средств вспомогательной терапии междисциплинарной группой врачей для облегчения контроля орган-специфических осложнений.
В Украине зарегистрированы два лекарственных средства для проведения ФЗТ у лиц с БФ: агалсидаза альфа и агалсидаза бета. Рекомендации относительно инициации ФЗТ у детей и взрослых в Проекте соответствуют приведенным выше (Hopkin R.J. et al., 2016; Ortiz A. et al., 2018).
Позитивному решению о начале ФЗТ способствует наличие органоспецифических критериев: кардиальных (гипертрофия сердца, нарушения сердечного ритма); неврологических (поражение белого вещества мозга, транзиторная ишемическая атака/инсульт, потеря слуха); ренальных (микроальбуминурия или протеинурия, хроническая болезнь почек стадии 2, 3а); нейропатической боли; симптомов со стороны ЖКТ.
В Проекте приведены противопоказания к инициации ФЗТ:
- относительные: беременность, лактация;
- абсолютные: прогнозируемая продолжительность жизни не более одного года; заболевания или сопутствующие состояния, при которых ФЗТ вряд ли повысит качество жизни, и состояния, при которых риск терапии выше пользы; постоянное тяжелое нейрокогнитивное нарушение; наличие антител IgE против агалсидазы.
В Проекте предложены варианты поддерживающей/симптоматической терапии и профилактических мероприятий, с которыми следует сочетать ФЗТ при наличии осложнений со стороны почек, сердца, ЖКТ, органов зрения и слуха, кожи, неврологических и других нарушений, связанных с БФ. Н.А. Пичкур отметила важность мониторинга осложнений со стороны внутренних органов у взрослых пациентов с БФ. В Проекте рекомендованы обследования и график мониторинга для различных органов и систем, в том числе, для оценки влияния ФЗТ.
Докладчик подчеркнула, что клиническая вариабельность БФ требует индивидуального подхода к лечению больных с учетом генотипа, пола, семейного анамнеза, фенотипа и степени тяжести специфических клинических симптомов у конкретного пациента.
***
По состоянию на 01.10.2018 г. в электронную базу украинского Центра орфанных заболеваний внесено 13 пациентов с БФ: 3 ребенка и 10 взрослых. Шесть из них получают ФЗТ, которая проводится препаратом агалсидаза бета (Фабразим®*), в том числе 4 пациента – за счет гуманитарной программы Sanofi Genzyme.
Подготовила Татьяна Ткаченко
* Препарат Фабразим® зареєстрований в Україні. Р.П. № UA/10306/01/01. Наказ МОЗ №939 від 05.12.2014 р.
** Диагностику редких заболеваний, в том числе болезни Фабри, по методу сухого пятна крови (DBS) бесплатно предоставляет ООО «Санофи-Авентис Украина».
Фермент-заместительная терапия при болезни Фабри: новые данные по эффективности разных дозировок
В ходе проспективного обсервационного исследования 112 пациентов (43 из них женского пола), которые были стабильными на агалсидазе бета по 1,0 мг/кг К2Н в течение не менее 12 месяцев, в нерандомизированном порядке разделили на три группы и проводили наблюдение в течение 53 (диапазон 38-57) месяцев:
- группа стандартного дозирования: 37 пациентов продолжали получать агалсидазу бета в дозировке 1,0 мг/кг/К2Н;
- переходная группа: 38 больных получали пониженную дозу агалсидазы бета и впоследствии переходили на агалсидазу альфа в дозе 0,2 мг/кг/К2Н или сразу же переходили на агалсидазу альфа по 0,2 мг/кг/К2Н и оставались на агалсидазе альфа в дозе 0,2 мг/кг/К2Н;
- группа повторного перехода: 37 пациентов снова переходили на агалсидазу бета в дозировке 1,0 мг/кг/К2Н после того, как получали агалсидазу альфа по 0,2 мг/кг/К2Н в течение не менее 12 месяцев.
Функция почек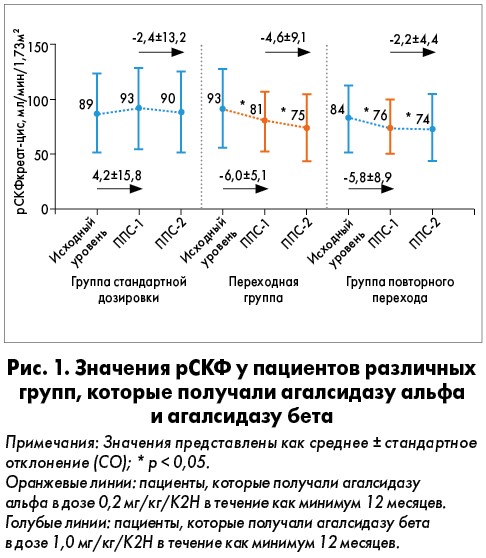
У пациентов из группы повторного перехода на агалсидазу бета в дозировке 1,0 мг/кг/К2Н наблюдалось снижение значений расчетной СКФ (рСКФ) (р<0,05) (рис. 1). Ежегодное снижение значений рСКФ от периода последующего наблюдения 1 (ППН-1) до периода последующего наблюдения 2 (ППН-2) составило 4,6 мл/мин/1,73 м2 в переходной группе по сравнению с 2,2 мл/мин/1,73 м2 – повторного перехода. В группе стандартного дозирования значения рСКФ оставались стабильными в течение всего ППН.
Гастроинтестинальные симптомы
Повторный переход на агалсидазу бета в дозировке 1,0 мг/кг/К2Н приводил к значительному снижению частоты случаев диареи (р<0,05). У пациентов из переходной группы чаще сообщалось о диарее во время первого долгосрочного ППН (р<0,05). В группе регулярных доз симптомы со стороны СКФ оставались стабильными в течение всего ППН.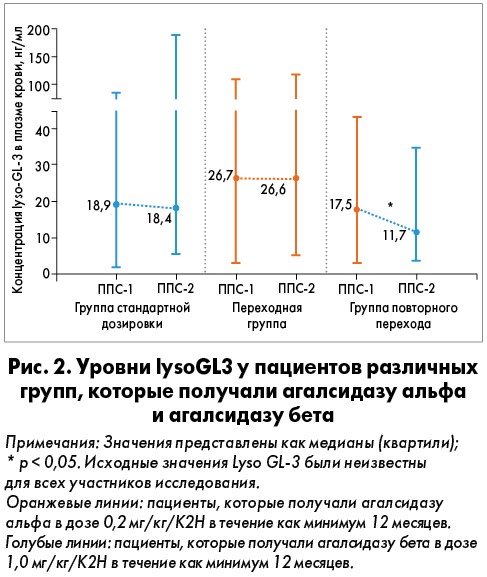
Уровни глоботриаозилсфингозина
В группе повторного перехода на агалсидазу бета по 1,0 мг/кг/К2Н отмечалось значительное снижение уровней глоботриаозилсфингозина (lyso-GL-3) (р<0,05) (рис. 2). Уровни lyso-GL-3 оставались стабильными между ППН-1 и ППН-2 в группе стандартного дозирования и переходной группе. У пациентов из переходной группы имели место высокие значения lyso-GL-3 во время ППН-1 (р<0,05 по сравнению с таковыми стандартного дозирования и повторного перехода).
Безопасность
Повторный переход на агалсидазу бета в дозировке 1,0 мг/кг/К2Н хорошо переносился и не приводил к клинически значимым инфузионным побочным реакциям. У троих (8%) пациентов в группе повторного перехода наблюдались незначительные инфузионные явления (незначительное повышение температуры тела и чрезмерная утомляемость).
Тематичний номер «Кардіологія, Ревматологія, Кардіохірургія» №1 (62), березень 2019 р.