


17 червня, 2020
Токсичні ускладнення імунотерапії
 Загальні аспекти застосування блокаторів імунних контрольних точок
Загальні аспекти застосування блокаторів імунних контрольних точок
Захворюваність та епідеміологія
Імунотерапія моноклональними антитілами (МоАТ), спрямована на цитотоксичний Т-лімфоцитасоційований антиген 4 (CTLA4), програмований білок клітинної смерті‑1 (PD‑1) і його ліганд (PD-L1), стає стандартом лікування для щоразу більшої кількості пацієнтів та супроводжується імовірним розвитком ускладнень. Частота токсичності змінюється залежно від імунної контрольної точки (immune checkpoint), на яку націлено лікування. Токсичність, зумовлену введенням інгібіторів імунних контрольних точок (ICPis), можна розділити на інфузійні реакції та небажані події, пов’язані з імунітетом (імНП), або небажані події, що становлять особливий інтерес. Будь-який орган чи тканина можуть бути уражені, хоча деякі імНП зустрічаються набагато частіше, ніж інші. Найчастіше імНП виникають з боку шкіри, товстої кишки, ендокринних органів, печінки та легенів. Інші імНП розвиваються дуже рідко, але можуть бути дуже серйозними, навіть летальними, наприклад, неврологічні розлади та міокардит.
Іпілімумабасоційована імунна токсичність
імНП, викликані іпілімумабом (анти-CTLA4) у дозі 3 мг/кг маси тіла, переважно 1 та 2 ступенів, фіксували у 60‑85% пацієнтів [1, 2], але у 10-27% пацієнтів розвивається токсичність 3-4 ступенів. У першому клінічному дослідженні III фази зафіксовано 2,1% випадків смерті, спровокованих прийомом іпілімумабу [1]. Початок проявів токсичності різниться, але зазвичай відзначається протягом перших 8-12 тижнів лікування, при цьому шкірні прояви часто виявляються першими. Ця токсичність є дозозалежною: при дозі 0,3 мг/кг відсутні небажані події (НП) 3-4 ступеня, тоді як при дозі 10 мг/кг частота проявів токсичності зростає до 30% [3]. При ад’ювантному застосуванні іпілімумабу 10 мг/кг з наступною підтримувальною дозою зареєстрована частка імНП 3-4 ступенів становила 41,6%, а 5 ступеня – 1,1% [4].
PD‑1/PD-L1-блокада та пов’язана з нею імунна токсичність
Найчастішою НП, спричиненою анти-PD‑1/PD-L1-терапією, є втома. Частота втоми, патогенез якої недостатньо вивчений у рамках досліджень окремих препаратів, становить 16-37% для анти-PD‑1 та 12-24% для анти-PD-L1 [5]. Тільки в невеликої частини пацієнтів втому можна асоціювати з гіпотиреозом. Токсичність високого ступеня, викликана анти-PD‑1-терапією (ніволумабом чи пембролізумабом), зустрічається рідше, ніж при застосуванні блокатора CTLA4 іпілімумабу.
Що стосується ніволумабу, будь-яка НП, пов’язана з лікуванням, була зафіксована у 74-85% пацієнтів, 3 та 4 ступеня – у 12-20% [2, 6, 7] хворих на метастатичну меланому, 58 та 7% відповідно – на місцевопоширений цисплатинрезистентний недрібноклітинний рак легені [8], 69 та 10% відповідно – на метастатичний цисплатинрезистентний неплоскоклітинний недрібноклітинний рак легені [9] та 79 і 19% відповідно – резистентну до інгібіторів тирозинкінази метастатичну нирковоклітинну карциному [10].
CTLA4- та PD‑1/PD-L1-препарати та пов’язана з ними імунна токсичність
Комбінована імунотерапія була схвалена лише у пацієнтів з метастатичною меланомою. НП, пов’язані з лікуванням, спостерігалися у 95% хворих. У 55% пацієнтів тяжкість цих НП була 3 ступеня або вищою [2]. Початок імунотоксичності 3-4 ступеня може бути різний як для монотерапії ніволумабом, так і для комбінованої імунотерапії, оскільки імНП можуть не тільки розвиватися раніше, як при комбінованій терапії, а й виникати через тривалий час.
Загальні аспекти небажаних подій, пов’язаних з імунітетом
Загалом імНП виникають досить рано, переважно протягом перших тижнів до 3 місяців після початку терапії блокаторами імунних контрольних точок. Однак вперше початок імНП був зафіксований через 1 рік після припинення лікування.
Роль біопсії тканин у діагностиці імунотоксичності не встановлена. У деяких рекомендаціях ідеться про біопсію тканин при токсичності вищого ступеня (3 та 4) щодо шкіри, шлунково-кишкового тракту, печінки, нирок, легенів, у разі виникнення сумнівів щодо етіології ускладнення та якщо лікування може бути змінено за результатом біопсії [15]. Загалом, коли біопсія проводиться за таких обставин, патоморфолог повинен бути повідомлений про конкретні причини проведення біопсії.
Відбір пацієнтів і базова оцінка
Перед початком лікування у пацієнтів слід оцінити схильність до розвитку імНП. Пацієнти з аутоімунним захворюванням в анамнезі або які активно лікуються від аутоімунного захворювання зазнають ризику погіршення аутоімунного захворювання під час імунотерапії [16]. Так само пацієнти, у яких виникли імНП на фоні лікування іпілімумабом, мають ризик розвитку імНП після лікування анти-PD‑1-препаратом і навпаки [16, 17]. Час між останньою дозою першого препарату та ініціацією наступного препарату може бути важливим, враховуючи тривалий період напіввиведення цих МоАТ.
Пацієнтів слід поінформувати про потенційні НП імунотерапії до початку лікування. Після розвитку імНП потрібно негайно провести клінічне обстеження хворого і вжити заходів для запобігання подальшому загостренню побічної реакції. У багатьох випадках, особливо найтяжчих, слід припинити імунотерапію, а для подолання токсичності необхідно призначити імуносупресивні або імуномодулювальні препарати, включаючи високі дози кортикостероїдів, а іноді антагоністи фактора некрозу пухлини, мікофенолат або такролімус, із подальшим обережним зменшенням імуносупресії. Тривале (>6 тижнів) лікування імуносупресивними препаратами або застосування інфліксимабу збільшує ймовірність розвитку опортуністичних інфекцій, тому профілактика пневмоцистних інфекцій має розглядатися відповідно до локальних рекомендацій. Важливо, що поки немає доказів впливу на клінічний результат у пацієнтів, які лікуються ICPis, імуносупресивних засобів, застосованих для корекції імунологічної токсичності [7, 18].
Шкірні прояви імунної токсичності
Захворюваність
НП з боку шкіри є одними з найчастіших у пацієнтів, які отримували МоАТ, що інгібують імунні контрольні точки, – CTLA4 (у 43-45% пацієнтів, які застосовували іпілімумаб) або PD‑1 (близько 34% хворих, котрі приймали ніволумаб і пембролізумаб) [1, 3, 19, 20] – і зазвичай розвиваються на початку лікування (протягом перших кількох тижнів).
Однак серйозні НП з боку шкіри зустрічаються рідко і зазвичай не потребують зниження дози або припинення лікування. Вітиліго як НП, пов’язана з імунотерапією, може бути асоційована з хорошим клінічним ефектом застосування анти-PD‑1 МоАТ у пацієнтів з меланомою [21].
Найчастішими шкірними проявами є висип, свербіж і вітиліго, але останнє спостерігається здебільшого у пацієнтів, які отримували лікування з приводу меланоми [20]. Повідомлялося про висип у близько 24% пацієнтів, які застосовували іпілімумаб, у близько 15% осіб, котрі отримували анти-PD‑1 МоАТ і в 40% – комбінацію іпілімумабу та ніволумабу. Однак висип 3 або 4 ступеня є рідкісним, із частотою <3% при монотерапії іпілімумабом або анти-PD‑1-препаратом і <5% при їх комбінації [2, 22]. Свербіж виявляли приблизно в 25-35% випадків застосування іпілімумабу, у 13-20% – анти-PD‑1 та 33% – у разі їх комбінації, але 3-4 ступеня лише в <2,5% [22]. Приблизно у 8% пацієнтів з меланомою, яких лікували анти-PD‑1 МоАТ [20] або комбінацією інгібіторів контрольних точок, відзначають вітиліго. У невеликому проспективному дослідженні вітиліго було виявлено у 25% пацієнтів, які отримували лікування пембролізумабом [21]. Цілком імовірно, що виникнення шкірних НП недостатньо ретельно зафіксовано в клінічних випробуваннях, оскільки зазвичай пацієнти систематично не проходять огляд дерматолога. У цьому дослідженні виникнення вітиліго було пов’язане з клінічним ефектом препарату. Розвиток вітиліго спостерігається переважно у хворих на меланому, яких лікували ICPis, але не при недрібноклітинному раку легені або раку нирки. Рідше повідомлялося про інші шкірні прояви токсичності ICPis: вогнищеву алопецію, стоматит, ксероз шкіри та фоточутливість, про загострення псоріазу, а також про псоріазоподібні або ліхеноїдні шкірні реакції у пацієнтів без жодного з таких захворювань в анамнезі [19, 23].
Гістопатологічно шкірні реакції можна розподілити на чотири групи [24]:
- запальні захворювання шкіри, що включають цілу низку змін, які відображають різні форми гострого, підгострого або хронічного запалення, пов’язаного з різноманітними епідермальними змінами, у тому числі псоріазоподібні чи ліхеноїдні шкірні реакції; часто спостерігають ліхеноїдні прояви хронічного дерматиту [25, 26];
- імунобульозні ураження шкіри, подібні до герпетиформного дерматиту чи бульозного пемфігоїда;
- альтерація кератиноцитів – хвороба Гровера [27]/акантолітичний дискератоз;
- імунна реакція, пов’язана з альтерацією меланоцитів (регресія невусів, акантолітичний дискератоз, пухлинний меланоз і вітиліго).
Діагностика та патоморфологія/молекулярна біологія
Коли у пацієнта, якого лікують ICPis, виникає НП з боку шкіри, насамперед необхідно виключити будь-яку іншу етіологію, наприклад, інфекцію, вплив іншого лікарського засобу або шкірні прояви інших системних захворювань. Далі ступінь тяжкості шкірних змін необхідно ретельно оцінити шляхом огляду, включаючи ділянки слизової оболонки, а також з’ясувати загальний стан пацієнта (лихоманка, збільшені лімфатичні вузли тощо), а при необхідності виконати аналіз крові, дослідження функції печінки та нирок. Це допоможе виключити можливість виникнення невідкладних дерматологічних станів, таких як медикаментозно зумовлений висип з еозинофілією та системними симптомами (DRESS), гострий фебрильний нейтрофільний дерматоз (Sweet syndrome), синдром Стівенса – Джонсона, або токсичний епідермальний некроліз. При таких загрозливих для життя станах (летальні випадки вже описані) лікування ICPis необхідно назавжди припинити, пацієнта госпіталізувати та негайно розпочати симптоматичне спеціалізоване лікування.
Щоб оцінити ступінь тяжкості НП з боку шкіри, зазвичай використовують класифікацію загальних термінологічних критеріїв побічних явищ (CTCAE).
Щодо макулопапульозного висипу, найчастішої шкірної НП, повязаної з застосуванням ICPis, у четвертій версії CTCAE подано таку класифікацію:
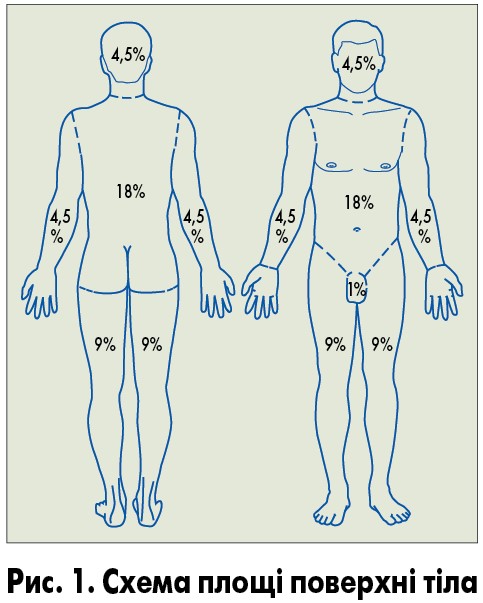 1 ступінь: макули/папули, що вкривають <10% площі поверхні тіла (ППТ; рис. 1), із симптомами або без них (наприклад, свербіж, жар, ущільнення);
1 ступінь: макули/папули, що вкривають <10% площі поверхні тіла (ППТ; рис. 1), із симптомами або без них (наприклад, свербіж, жар, ущільнення);
2 ступінь: макули/папули, що вкривають 10-30% ППТ, із симптомами або без них; обмеження щоденної активності;
3 ступінь: макули/папули, що вкривають >30% ППТ, із супутніми симптомами або без них; обмеження здатності до самообслуговування та щоденної активності;
4 ступінь: папулопустульозний висип, пов’язаний із небезпечною для життя суперінфекцією; синдром Стівенса – Джонсона та бульозний дерматит, що уражають >30% ППТ; потребує лікування у відділенні реанімації та інтенсивної терапії.
Інструментальне визначення щоденної активності або здатності до самообслуговування доцільне для оцінювання ступеня тяжкості НП, а також її впливу на життя пацієнта. Однак той факт, що при ураженні >30% ППТ висип автоматично оцінюється як подія 3 ступеня, є дискусійним. Дійсно, коли висип є дифузним, але легким і не супроводжується будь-якими додатковими симптомами, оцінити ці зміни як НП 2 ступеня більш доцільно.
П’ята версія CTCAE подасть відповіднішу класифікацію шкірних НП.
Лікування висипу
У випадку шкірних НП 1 ступеня, таких як висип та/або свербіж, лікування ICPis можна продовжувати (рис. 2). Можна використовувати місцеві пом’якшувальні засоби, пероральні антигістамінні препарати та/або середньої сили кортикостероїди місцевої дії. У разі шкірних проявів 2 ступеня лікування ICPis можна продовжувати, але пацієнтів слід щотижня оглядати для перевірки клінічної ситуації. Якщо шкірні НП не зникли, лікування слід припинити доти, доки шкірні НП не стануть 1 ступеня. Симптоматичне лікування складається з місцевих пом’якшувальних засобів, пероральних антигістамінних препаратів і місцевих кортикостероїдів середньої та високої сили дії. Шкірні НП 3 ступеня також потребують негайного припинення ICPis, поки їх вираженість не знизиться до 1 ступеня. Лікування включає місцеві пом’якшувальні засоби, пероральні антигістамінні препарати та місцеві кортикостероїди високої сили дії (II, B). Системні кортикостероїди у дозі 0,5-1 мг/кг можна призначати залежно від вираженості симптомів. У рідкісних випадках шкірних НП 4 ступеня лікування ICPis слід перервати, а пацієнтів негайно помістити під нагляд дерматолога. Лікування складається з внутрішньовенного введення (метил)преднізолону у дозі 1-2 мг/кг із зниженням дози після нормалізації токсичності (II, B).
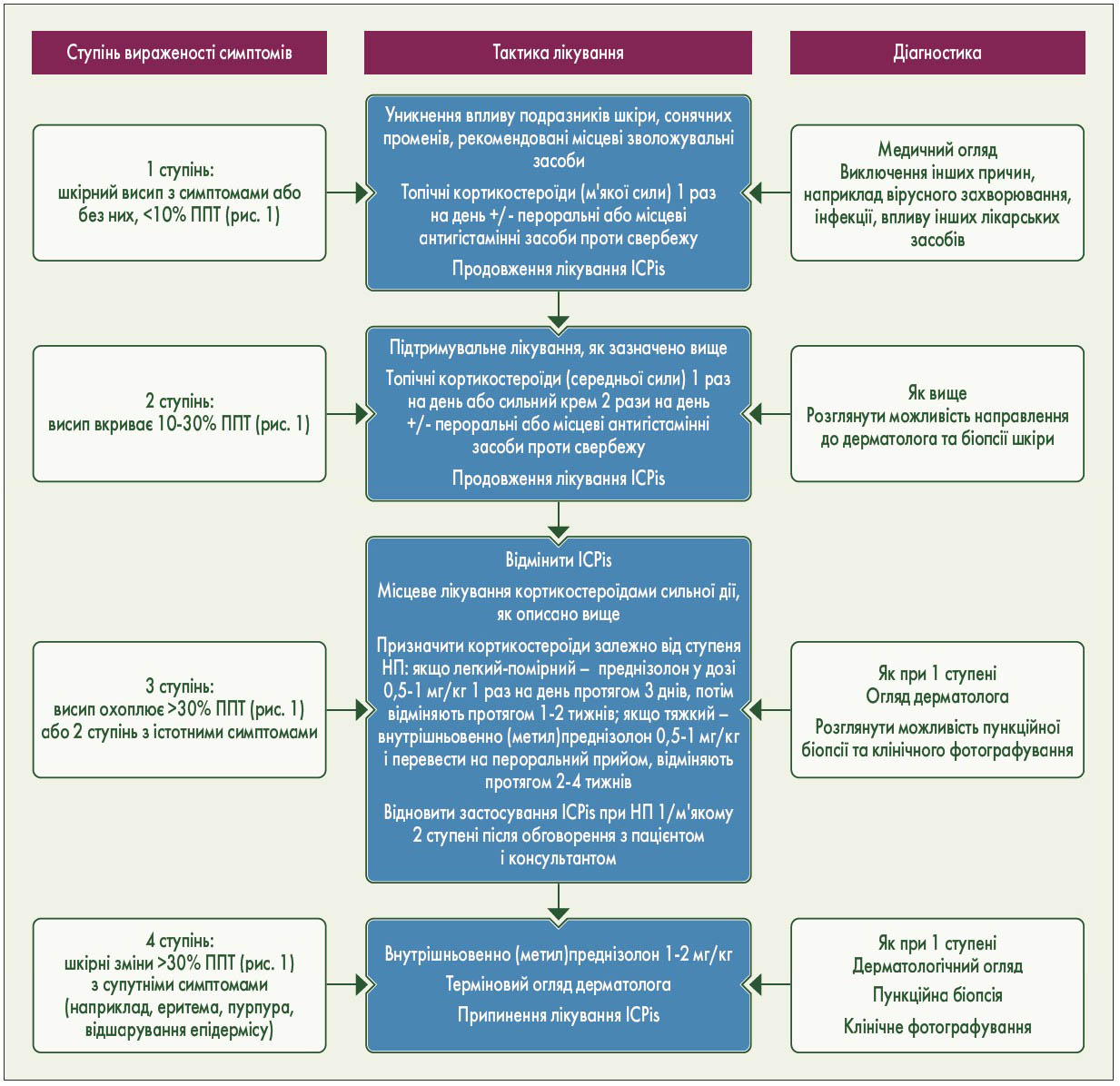 Рис. 2. Пов’язана з ICPis токсичність: лікування шкірного висипу/токсичності. НП з боку шкіри: (i) найпоширеніші – еритема, макулопапульозний та пустулопапульозний висип; (ii) рідкісні: токсичний епідермальний некроліз, синдром Стівенса – Джонсона та DRESS; (iii) васкуліт також може бути представлений висипом типу пурпури
Рис. 2. Пов’язана з ICPis токсичність: лікування шкірного висипу/токсичності. НП з боку шкіри: (i) найпоширеніші – еритема, макулопапульозний та пустулопапульозний висип; (ii) рідкісні: токсичний епідермальний некроліз, синдром Стівенса – Джонсона та DRESS; (iii) васкуліт також може бути представлений висипом типу пурпури
Ендокринопатії, пов’язані з імунною токсичністю
Дисфункція щитоподібної залози
Хоча розлади функції щитоподібної залози спостерігаються досить часто у пацієнтів, які отримували імунотерапію, таку як цитокіни інтерлейкін‑2 та інтерферони I типу, їх частота значно зросла з моменту впровадження ICPis у клінічну практику. Повідомлялося про гіпер- та гіпотиреоз, хоча гіпотиреоїдні стани зустрічаються частіше, ніж гіпертиреоз. Останній часто минущий і може передувати гіпотиреозу. Досі мало відомо про патогенез порушень щитоподібної залози після прийому ICPis. Вважається, що він виникає за участі Т-лімфоцитів.
Дисфункція щитоподібної залози найчастіше розвивається після лікування анти-PD‑1/PD-L1-препаратами або комбінацією анти-CTLA4-препаратів і блокаторів PD‑1/PD-L. Застосування іпілімумабу (3 мг/кг) викликає порушення функції щитоподібної залози в 1-5% хворих [1, 2], але збільшення дози (10 мг/кг) супроводжується зростанням частоти виникнення НП з боку щитоподібної залози (до 10%) [4]. Лікування анти-PD‑1- (пембролізумаб або ніволумаб) чи анти-PD-L1-препаратами (атезолізумаб) провокує дисфункцію щитоподібної залози у 5-10% (незалежно від типу пухлини) [6, 10, 12]. При комбінованій імунотерапії (іпілімумаб 3 мг/кг + ніволумаб 1 мг/кг) частота порушень функції щитоподібної залози збільшується до 20% [2]. Тяжкість цих НП рідко перевищує 2 ступінь. У більшості випадків дисфункцію щитоподібної залози виявляють за допомогою рутинного аналізу крові (тиреотропний гормон – ТТГ та вільний тироксин – FТ4); їх слід проводити перед кожною інфузією або принаймні 1 раз на місяць (у разі інфузій через 2 тижні).
Лікування
Призначення замісної терапії гормоном щитоподібної залози слід розглядати у разі втоми або інших скарг, які можна асоціювати з гіпотиреозом (IV-V, B). Пацієнтам із симптомами, особливо гіпертиреозу, слід розпочати лікування блокаторами бета-адренорецепторів (пропранололом або атенололом; IV-V, B). Рідко застосовують карбімазол або кортикостероїди. У цих випадках лікування ICPis слід припинити до усунення симптомів. Замісна гормональна терапія зазвичай є тривалою (рис. 3).
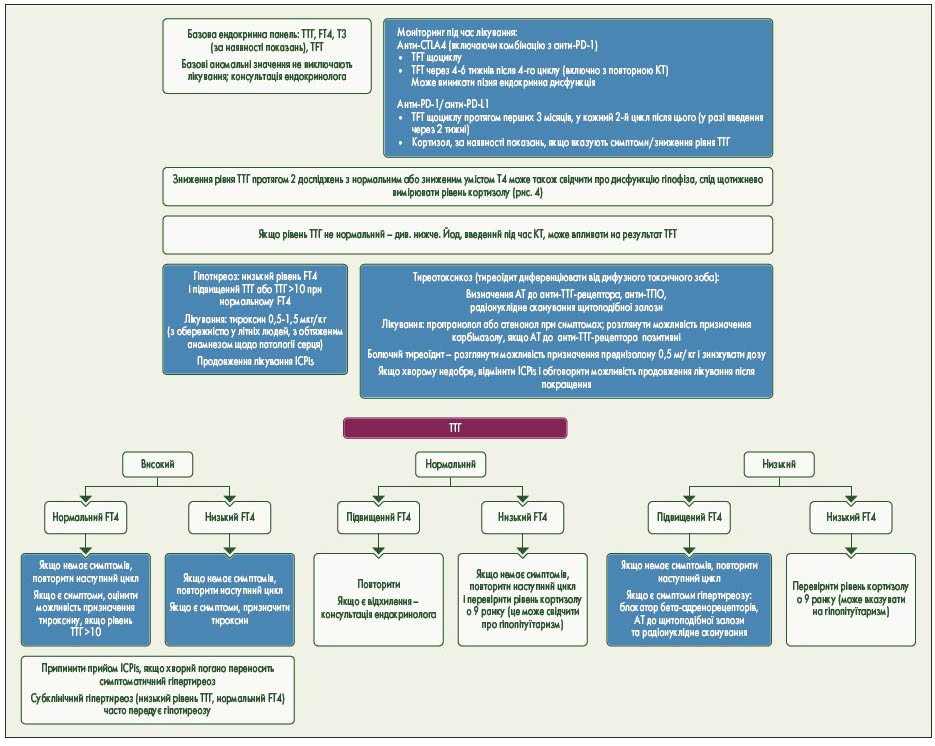 Рис. 3. Пов’язана з ICPis токсичність: моніторинг і лікування дисфункції щитоподібної залози. AТ – антитіло; КТ – комп’ютерна томографія; Т3 – трийодтиронін; TFT – тест функції щитоподібної залози; ТПО – тиреоїдна пероксидаза
Рис. 3. Пов’язана з ICPis токсичність: моніторинг і лікування дисфункції щитоподібної залози. AТ – антитіло; КТ – комп’ютерна томографія; Т3 – трийодтиронін; TFT – тест функції щитоподібної залози; ТПО – тиреоїдна пероксидаза
Гіпофізит
До впровадження у клінічну практику анти-CTLA4-терапії гіпофізит (запалення передньої частки гіпофіза) був надзвичайно рідким. Повідомляється про виникнення гіпофізиту при використанні іпілімумабу у дозах 3 та 10 мг/кг та комбінації іпілімумабу та ніволумабу відповідно в 1; 16 та 8% пацієнтів [1, 2, 4]. Гіпофізит дуже рідко розвивався у пацієнтів, які отримували анти-PD‑1- та анти-PD-L1-терапію [29].
Пацієнти можуть мати різні скарги. Головний біль та порушення зору вимагають негайної оцінки та диференціації від церебральних метастазів, лептоменінгеальної хвороби, цереброваскулярних захворювань та гіпофізиту. При магнітно-резонансній томографії (МРТ) головного мозку може бути видно набряк або збільшення гіпофіза. Часто одночасно низький рівень ТТГ, адренокортикотропного гормону (АКТГ) в крові та/або фолікулостимулюючого (ФСГ)/лютеїнізуючого гормону (ЛГ) вказують на гіпофізит як на найбільш вірогідний діагноз. Маніфестація хвороби може бути зумовлена гіпотиреозом та/або гіпокортизолізмом (недостатністю надниркових залоз) та низьким рівнем тестостерону.
Лікування
Після підтвердження діагнозу при гіпофізиті 2 ступеня або вищого лікування ICPis слід припинити, а гормонозамісне лікування – негайно розпочати (V, B). У разі головного болю та інших неврологічних проблем слід розпочати застосування високих доз кортикостероїдів; однак, імовірно, високі дози кортикостероїдів не протидіють гормональній недостатності, зумовленій анти-CTLA4-лікуванням. У більшості випадків прийом ICPis можна продовжити. Більшості пацієнтів потрібна тривала гормонозамісна терапія (рис. 4).
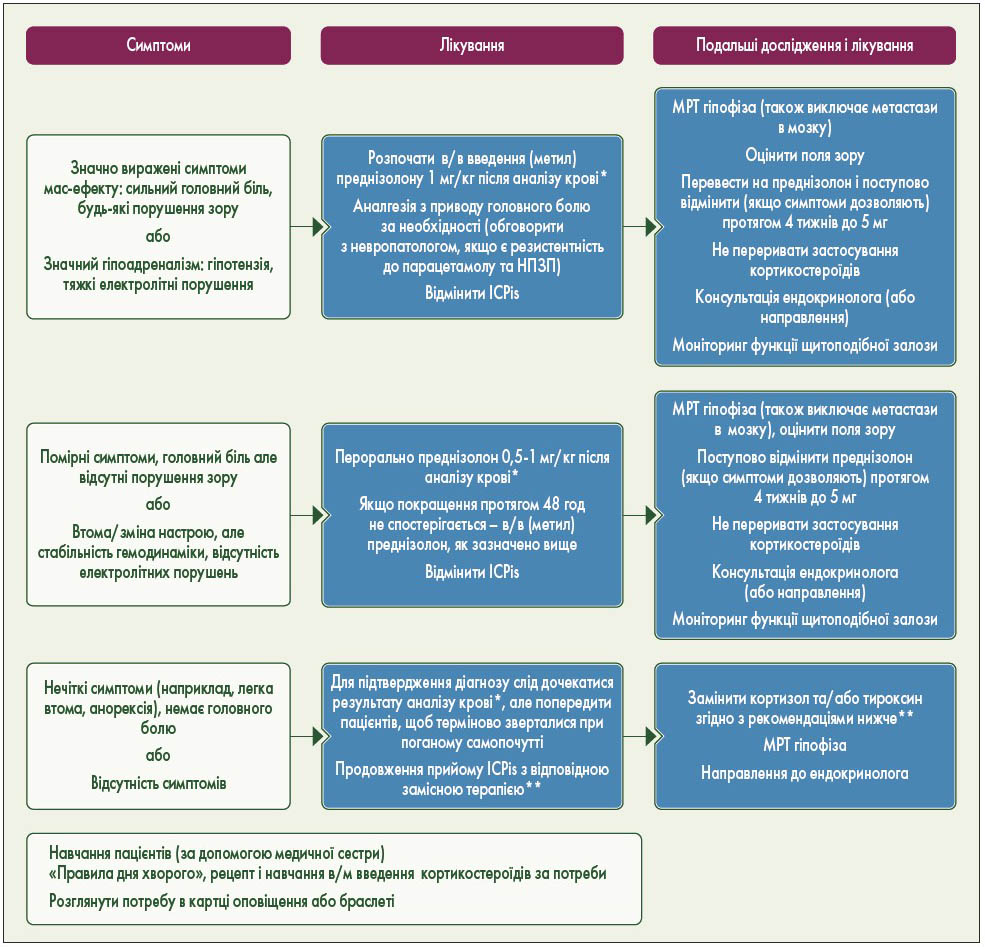
Рис. 4. Пов’язана з ICPis токсичність: діагностика і лікування гіпофізиту
*Аналіз крові: о 9 ранку із визначенням рівня кортизолу (або в інший час, якщо хворому погано і лікування не може бути відкладено), АКТГ, ТТГ/FТ4, ЛГ, ФСГ, естрадіолу (у жінок у пременопаузі), тестостерону у чоловіків, інсуліноподібного фактора росту‑1, пролактину. Замісна терапія мінералокортикоїдами рідко необхідна при гіпопітуїтаризмі.
** Початкові рекомендації щодо замісної терапії кортизолу та тиреоїдних гормонів:
- Якщо рівень кортизолу о 9 ранку <250 або в інший час <150 і невиражені симптоми:
- замініть гідрокортизоном 20/10/10 мг;
- якщо результат TFT нормальний, спочатку моніторинг 1-2 тижні (завжди замінюйте кортизол за 1 тиждень до початку терапії тироксином);
- Якщо знижується рівень ТТГ +/- низький FT4:
- розгляньте необхідність заміни тироксину (орієнтовно 0,5-1,5 мг/кг) з урахуванням симптомів +/- визначення рівня кортизолу о 9 ранку щотижня;
- див. Рекомендації щодо щитоподібної залози для отримання додаткової інформації щодо інтерпретації аномального ТТГ/Т4.
Цукровий діабет 1 типу
De novo діабет, індукований лікуванням ICPis, виникає з низькою частотою (<1%). Цукровий діабет (ЦД) частіше зустрічається при блокаді PD‑1 та PD-L1 (або комбінованій імунотерапії), ніж при застосуванні іпілімумабу [31]. Шлях PD‑1 відіграє роль у розвитку аутоімунного ЦД, оскільки блокада PD‑1/PD-L1 запускає виникнення на моделях тварин ЦД 1 типу, опосередкованого специфічними CD8 T-лімфоцитами. Однак захворюваність на ЦД 1 типу може підвищитися унаслідок лікування більшої популяції пацієнтів анти-PD‑1- або анти-PD-L1-препаратами.
Рекомендується проводити моніторинг у пацієнтів, які отримують ICPis, з метою виявлення ЦД шляхом визначення рівня глюкози в крові. Може виникати ЦД як 1, так і 2 типів. Навіть у пацієнтів із ЦД 2 типу може розвинутися кетоацидоз – нечаста, але небезпечна для життя подія, лікування при якій призначають відповідно до стандартних рекомендацій (I, A). Чи може лікування високими дозами кортикостероїдів запобігти повній втраті бета-клітин на острівцях Лангерганса, не зрозуміло. Кортикостероїди радше негативно впливатимуть на контроль діабету у цих пацієнтів.
С-пептид та антитіла проти декарбоксилази глутамінової кислоти та острівцевих клітин слід вимірювати, щоб розрізняти ЦД 1 та 2 типів. Після стабілізації стану пацієнта замісною інсулінотерапією може бути розглянута можливість відновлення лікування ICPis.
Гепатотоксичність, пов’язана з лікуванням
Захворюваність
Гепатит зустрічається у 5-10% (з них 1-2% – це НП 3 ступеня) пацієнтів під час монотерапії іпілімумабом, ніволумабом і пембролізумабом у дозволених дозах та у 25-30% (з них 15% – це НП 3 ступеня) тих, хто отримував комбінацію іпілімумабу 3 мг/кг і ніволумабу 1 мг/кг [2, 12].
Діагностика
Усіх пацієнтів, які проходять терапію ICPis, слід перевіряти на ознаки та симптоми гепатиту, рівень сироваткових трансаміназ та білірубіну перед кожним циклом лікування. Гепатит, як правило, має безсимптомний перебіг і виявляється при звичному контролі показників крові. Якщо розвивається гепатит, слід виключити причини, пов’язані із захворюванням, супутнє вживання наркотиків і алкоголю, інфекційні причини, зокрема вірусні гепатити. Однак початок терапії, за її необхідності, не слід відкладати до отримання результатів серологічних досліджень, якщо немає іншої видимої причини.
Біопсія печінки може розглядатися як допоміжний метод диференційної діагностики більш тяжких гепатитних реакцій [15]. Найчастіше повідомлялося про лобулярний гепатит, який не відрізняється від аутоімунного [32, 33]; у більшості випадків він є панлобулярним, але запалення може бути обмежене 3-ю зоною. Наявність синусового гістіоцитозу та ендотеліїту центральних вен може допомогти виявити пов’язане з іпілімумабом запалення. У рідкісних випадках можливе запалення портального тракту та холангіт або зміни, які не відрізняються від неалкогольного стеатогепатиту.
Лікування
У разі помірного (2 ступеня) підвищення рівня трансаміназ або загального білірубіну слід припинити терапію ICPis, а рівень трансаміназ і білірубіну вимірювати 2 рази на тиждень. У разі стійкого підвищення 2 рівня довше 1-2 тижнів (після виключення інших причин) слід призначити кортикостероїди в дозі 1 мг/кг на добу – (метил)преднізолон або еквівалент. Після покращення результатів терапію ICPis можна відновити після редукції дози кортикостероїдів. Якщо ситуація погіршується або не відбувається покращення, незважаючи на застосування кортикостероїдів, слід збільшувати дозу кортикостероїдів – (метил)преднізолону або його еквівалентів – до 2 мг/кг на добу, а терапію ICPis припинити (IV-V, B). При підвищенні рівня трансаміназ або загального рівня білірубіну до 3-4 ступеня терапію ICPis слід припинити, а введення кортикостероїдів – (метил)преднізолону або еквівалентів – починати з дози 1-2 мг/кг на добу. Якщо протягом 2-3 днів немає відповіді на кортикостероїди, слід додати мікофенолату мофетил у дозі 1000 мг 2 рази на добу (IV-V, B) [34]. Консультація гепатолога та розгляд потреби біопсії печінки (див. вище) рекомендується у разі рефрактерності до кортикостероїдів та мікофенолату мофетилу (IV-V, B). Імуносупресивна терапія третьої лінії недостатньо визначена, але було зареєстровано успішне застосування антитимоцитарного глобуліну у разі гепатиту, спричиненого іпілімумабом, резистентного до кортикостероїдів та мікофенолату мофетилу. Ще одним варіантом імуносупресивної терапії третьої лінії є такролімус. Інфліксимаб не рекомендується застосовувати для лікування гепатиту, пов’язаного з імунотоксичністю (рис. 5).
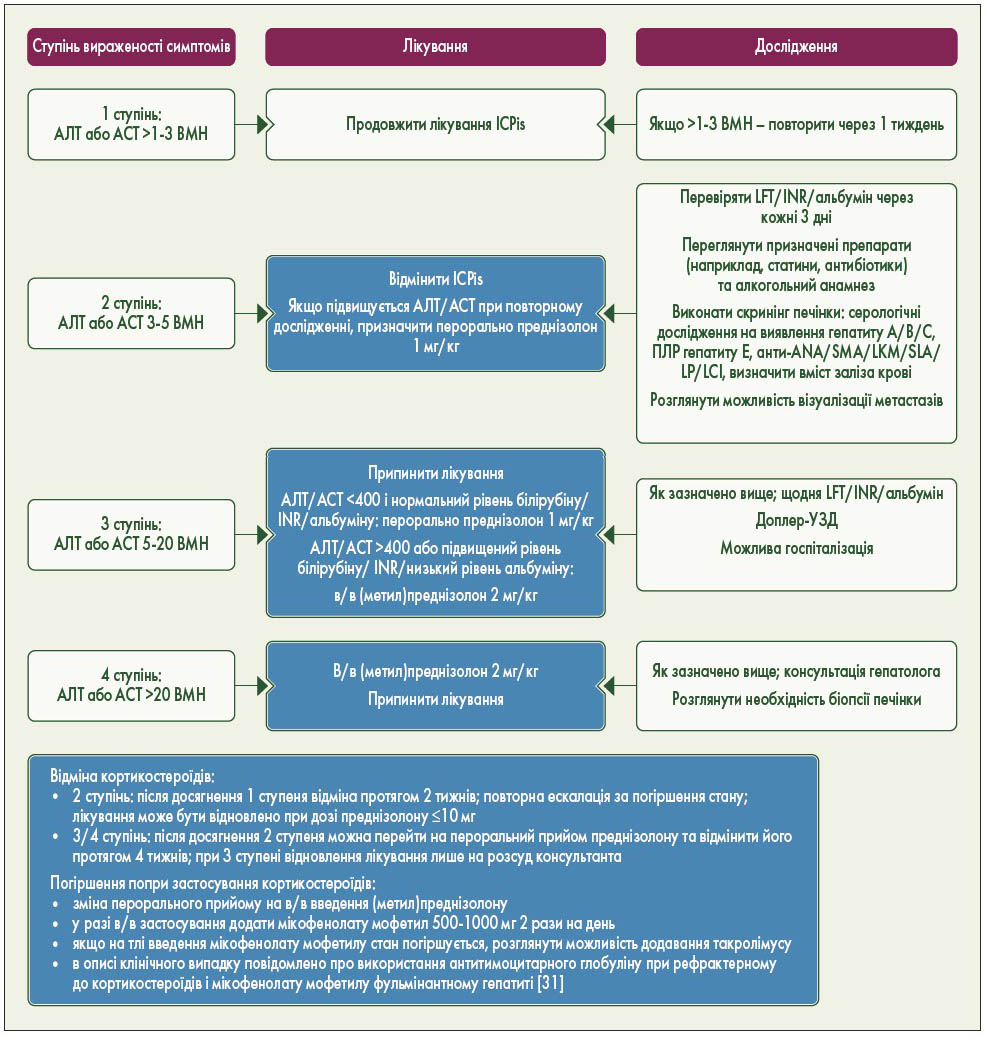
Рис. 5. Пов’язана з ICPis токсичність: лікування гепатиту
АЛТ – аланінамінотрансфераза; АСТ – аспартатамінотрансфераза; ANA – антиядерні АТ; INR – міжнародне нормалізоване співвідношення протромбінового часу; LCI – індекс кліренсу легенів; LFT – дослідження функції печінки; LKM – мікросомальні АТ до печінки/нирок; ПЛР – полімеразна ланцюгова реакція; SLA/LP – розчинний антиген печінки/антитіло до печінки та підшлункової залози; SMA – аутоантитіла до гладеньких м’язів; ВМН – верхня межа норми; УЗД – ультразвукове дослідження
Зазвичай при відповідному лікуванні гепатиту одужання настає протягом 4-6 тижнів, але якщо він не припиняється, слід ще раз проаналізувати інші можливі причини, а також, при потребі, повторити початкові діагностичні заходи, особливо беручи до уваги можливий супутній прийом інших гепатотоксичних препаратів (у тому числі засобів рослинного походження та тих, що продаються без рецепта) і реактивацію цитомегаловірусної інфекції.
Продовження у наступному номері.
Список літератури знаходиться в редакції.
Haanen J.B.A.G. et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology 28 (suppl. 4): i119-i142, 2017. DOI:10.1093/annonc/mdx225.
Переклав з англ. Назар Лукавецький
Тематичний номер «Онкологія, Гематологія, Хіміотерапія» № 2 (63) 2020 р.