


6 травня, 2023
Рідкісні причини акушерських і аномальних маткових кровотеч у жінок: діалог акушера та гематолога
За матеріалами конференції
Зважаючи на високу медичну значущість проблеми аномальних маткових кровотеч (АМК) та акушерських кровотеч (АК) у жінок, актуальним є мультидисциплінарний підхід до її вирішення. І хоча кровотечі традиційно вважаються акушерсько-гінекологічною проблемою, залучення до лікувально-діагностичного процесу лікаря-гематолога дозволяє виявити у пацієнток рідкісні (генетичні та аутоімунні) причини кровотеч і забезпечити їх подальше ефективне лікування. Тому програма вебінару, що відбувся 24 червня 2022 року і був присвячений цій актуальній міждисциплінарній темі, включала діалог двох експертів із різних галузей, яких об’єднала спільна проблема — менеджмент пацієнток із АМК та АК.
Погляд акушера-гінеколога на проблему АМК і АК висвітлила експерт Комітету з біоетики Ради Європи, доповідач комітету з гендерних питань, завідувач відділення акушерських проблем екстрагенітальної патології ДУ «Інститут педіатрії, акушерства і гінекології ім. академіка О.М. Лук’янової НАМН України», доктор медичних наук, професор Юлія Володимирівна Давидова.
Другим експертом, який розглянув зазначену проблему з точки зору лікаря-гематолога, виступила голова Асоціації гематологів України, завідувач Центру гематології та трансплантації кісткового мозку КНП КОР «Київський міський клінічний онкологічний центр», кандидат медичних наук Ірина Радомирівна Гартовська.
 Вебінар розпочався із презентації складного клінічного випадку пацієнтки, яка звернулася зі скаргами на рясні кров’яні виділення та біль у місці розрізу (кесарів розтин) через 12 міс після пологів. Наявні у хворої симптоми свідчили про розвиток патологічного процесу, що потребував проведення диференціальної діагностики, визначення діагнозу та призначення відповідного лікування.
Вебінар розпочався із презентації складного клінічного випадку пацієнтки, яка звернулася зі скаргами на рясні кров’яні виділення та біль у місці розрізу (кесарів розтин) через 12 міс після пологів. Наявні у хворої симптоми свідчили про розвиток патологічного процесу, що потребував проведення диференціальної діагностики, визначення діагнозу та призначення відповідного лікування.
Клінічний випадок 1
Пацієнтка Г., 29 років, звернулася з питання планування вагітності. З анамнезу: вагітність – перша, пологи — перші (кесарів розтин через клінічну невідповідність голівки плода тазу матері, обструктивні пологи).
Скарги: через 12 міс після пологів наявні рясні кров’яні виділення, біль у місці розрізу.
Лабораторні дослідження: подовжений час згортання крові, активний частковий тромбопластиновий час (АЧТЧ) — 69,65 сек.
Враховуючи скарги та результати лабораторних досліджень, першочерговим при менеджменті даної пацієнтки є збір анамнезу, який включає відповіді хворої на три ключові питання:
- яким є перебіг періоду після стоматологічних процедур?
- чи легко виникають синці при незначному травмуванні?
- який анамнез пубертатного періоду та сімейний анамнез?
Зі слів жінки, у дитинстві вона часто відзначала синці, тривалі кровотечі з ран і садна шкіри, а також спонтанну кровоточивість ясен і менорагії. Подібна історія синців та надмірної кровоточивості мала місце і в її брата. Що стосується матері пацієнтки, то, з її слів, мати має негативний анамнез щодо тенденцій до кровотеч, безпроблемний акушерський та менструальний анамнези.
Після проведення ультразвукового дослідження черевної порожнини та органів таза було виявлено: гіпоехогенне утворення 10-12 см3 спереду від матки до передньої стінки матки та гіпоехогенне утворення у внутрішньом’язовій площині, що вказувало на гематому.
Хворій було призначено терапію, яка включала ін’єкції вітаміну К, пероральний етамзилат, транексамову кислоту внутрішньовенно та три дози свіжозамороженої плазми. Призначена терапія значно покращила клінічні симптоми. У подальшому пацієнтці було призначено препарат транексамової кислоти.
Незважаючи на те що хвора отримала відповідне лікування, яке покращило її загальний стан, менеджмент на цьому не завершився, оскільки залишався під питанням остаточний діагноз захворювання. Вищезгадані симптоми, наявні у пацієнтки, могли бути пов’язані з трьома діагнозами. Так, рясна кровотеча могла бути спричинена хворобою Гоше, хворобою фон Віллебранда (Von Willebrand disease – VWD) та розвитком інгібіторної форми гемофілії. Враховуючи клінічний і сімейний анамнез пацієнтки, а також результати лабораторного дослідження, їй було встановлено діагноз VWD.
Найбільш поширені причини патології системи згортання крові у жінок загалом можна класифікувати на дві групи: вроджені й набуті розлади системи гемостазу. Перша група включає спадкові коагулопатії, зокрема VWD, носійство гена гемофілії (при спонтанній мутації в обох хромосомах) та дисфункцію тромбоцитарної ланки гемостазу (хвороба Гланцмана). Набута патологія системи згортання крові представлена набутою гемофілією, ідіопатичною тромбоцитопенічною пурпурою та комбінованою недостатністю факторів згортання (захворювання печінки).
VWD названа на честь її першовідкривача Еріка Адольфа фон Віллебранда, фінського педіатра, який уперше описав захворювання у 1926 році. Пацієнткою, лікування якої призвело до відкриття спадкового розладу коагуляції, що зараз має назву VWD, була п’ятирічна дівчинка, яка проживала на острові Еланд і була доставлена до госпіталю Діконес м. Гельсінкі в 1924 році та обстежена доктором Віллебрандом. Лікар обстежив 66 членів її сім’ї й дійшов висновку, що це не описаний до того часу розлад системи згортання крові, відмінний від уже відомої на той час гемофілії. Даний розлад характеризувався наявністю кровотеч зі слизових оболонок, аутосомним типом успадкування, подовженим часом кровотечі за методом Дюка та нормальним згортанням крові.
Наразі відомо, що VWD – найпоширеніший спадковий розлад системи гемостазу, розповсюдженість якого у загальній популяції становить 1-2%. Більшість випадків захворювання є недіагностованими, оскільки найчастіше специфічні симптоми відсутні. Натомість поширеність симптомної VWD у 10-100 разів нижча і становить 0,1-0,01% у популяції. Частота діагностики різниться залежно від підтипів VWD, при цьому значна частка пацієнтів залишаються недостатньо лікованими. Примітно, що хоча VWD уражає обидві статі, вища частота захворюваності реєструється серед жінок. Жінки непропорційно до чоловіків частіше страждають у репродуктивному віці через рясні менструальні кровотечі та пологи. В останні роки відзначається покращення обізнаності медичного персоналу щодо цієї патології, що, відповідно, позначається й на діагностиці VWD: про це свідчить «помолодшання» хворих – вік понад 50% пацієнтів складає <20 років.
Клінічно VWD проявляється кровотечами із чотирьох основних ділянок: порожнина носа й рота, шлунково-кишковий тракт, суглоби та матка.
Алгоритм діагностики VWD включає три послідовні кроки: збір клінічного анамнезу, проведення диференціальної діагностики та встановлення діагнозу. Так, початок діагностики VWD має бути зосереджений на клінічній історії пацієнта, зокрема на оцінці сімейної історії кровотеч, а також особистого анамнезу щодо надмірних кровотеч протягом усього життя.
Наступний етап передбачає проведення диференціальної діагностики з іншими захворюваннями, які можуть супроводжуватися клінічною картиною кровотеч. Зокрема, він передбачає, що клініцист має зосередитися на стратегії виключення інших поширених причин кровотечі. Цей крок включає проведення лабораторного скринінгу, а саме загального аналізу крові з обов’язковим визначенням кількості тромбоцитів, дослідження мазка периферичної крові, вимірювання протромбінового часу (ПТЧ), АЧТЧ і часу кровотечі. При цьому важливо розуміти, що проведення коагулограми з наступним визначенням АЧТЧ є критично важливим для оцінки подальшої стратегії менеджменту пацієнток із кровотечами, оскільки більш традиційні лабораторні показники, такі як ПТЧ і міжнародне нормалізоване відношення (МНВ), не будуть інформативними. Після проведення всіх вищеперерахованих лабораторних досліджень і виключення всіх імовірних причин кровотечі рекомендовано визначити наявність/відсутність у хворої дефіциту факторів згортання крові, зокрема у жінок важливо оцінити фактор фон Віллебранда (Von Willebrand factor – VWF) – білок плазми крові, зниження активності або кількості якого є діагностичним маркером VWD. У разі відсутності дефіциту VWF рекомендовано розглянути інші причини кровотечі, зокрема оцінити наявність патології тромбоцитів, їх агрегацію тощо.
Третій крок менеджменту пацієнтки з підозрою на VWD включає постановку діагнозу. Так, наявність підтвердженого дефіциту VWF є діагностичним маркером VWD. Подальші дії передбачають визначення типу VWD: частковий кількісний дефіцит (тип 1), якісний дефіцит (тип 2) та повний дефіцит (тип 3) VWF. Саме цей етап викликає значні труднощі у клініцистів. Установлено, що тип 3 та більшість варіантів типу 2 VWD діагностуються відносно легко. Натомість діагностика VWD типу 1 є проблематичною, оскільки розрізняти доброякісні поліморфізми і патогенні мутації складно. Ще одним питанням, яке також ускладнює діагностування VWD типу 1, є той факт, що для підтвердження або виключення підозрюваного діагнозу може знадобитися кілька досліджень.
Після підтвердження діагнозу VWD перед лікарем виникає цілком логічне питання – які стратегії терапії на сьогодні є найбільш ефективними? Згідно із сучасними рекомендаціями, у доробку клініцистів наявні три загальні стратегії лікування VWD: сприяння активації гіперкоагуляційного синдрому, заміщення дефіцитного VWF та сприяння гемостазу й загоєнню ран.
Завдяки успіхам сучасної медицини існує значна кількість варіантів лікування гематологічних захворювань, які раніше вважалися вироком для пацієнта. Сьогодні відомо чимало препаратів, показаних для профілактики й лікування клінічних симптомів VWD. Так, при легкому перебігу VWD можливе застосування активаторів гіперкоагуляційного синдрому, зокрема препаратів десмопресину, механізм дії яких пов’язаний із викидом усіх факторів згортання крові з депо. Як наслідок, розвивається гіперкоагуляційний синдром, який і приводить до зупинки кровотечі. При тяжкому перебігу VWD та значних кровотечах показана замісна терапія відповідним дефіцитним фактором. Утім в Україні рекомбінантний VWF не зареєстрований, тому загальні підходи до лікування хворих із VWD передбачають застосування замісної терапії плазмовим VIII фактором згортання крові (FVIII) із високим вмістом VWF. Дана стратегія заснована на тому факті, що FVIII та VWF пов’язані між собою у плазмі крові, тому, як правило, усі плазмові фактори крові містять і VWF у тій чи іншій концентрації. Наразі в арсеналі вітчизняних клініцистів є більш концентровані щодо вмісту VWF плазмові FVIII, які й рекомендовані для лікування VWD. Третьою стратегією менеджменту VWD є застосування місцевої терапії, зокрема транексамової кислоти.
Гемофілія — рідкісне спадкове захворювання системи гемостазу
Зі спадкових порушень системи гемостазу найвідомішим є гемофілія – Х-зчеплене захворювання системи гемостазу, що характеризується зниженням або порушенням синтезу факторів згортання крові: FVIII при гемофілії типу А та IX фактора (FIX) при гемофілії типу В. Загальновідомо, що гемофілія успадковується за рецесивним типом, пов’язаним зі статтю. Ген, який відповідає за синтез FVIII і FIX, міститься у X-хромосомі, внаслідок чого на гемофілію хворіють виключно чоловіки, натомість як жінки можуть бути лише носіями мутантного гена. Останнім часом розуміння патофізіології гемофілії значно розширилося. І сьогодні жінок, які є носіями мутантного гена, прирівнюють до чоловіків, хворих на гемофілію, оскільки, хоча у жінок-носіїв рівень FVIII знаходиться в нижній межі норми, при будь-яких оперативних і стоматологічних процедурах, при пологах вони можуть мати високий ризик кровотечі. Згідно з міжнародними рекомендаціями, наразі жінок-носіїв гена гемофілії забезпечують замісною гормональною терапією фактором згортання крові. Ця норма стосується виключно лікування, тоді як профілактичної терапії, на відміну від чоловіків, жінки цієї групи не потребують.
Крім того, сьогодні існує також проблема спонтанних мутацій. Згідно із сучасними даними, близько 30% усіх випадків гемофілії є результатом спонтанних генетичних мутацій. Відомо, що гемофілія успадковується через Х-хромосому з мутацією генів F8 та F9. Саме ці два гени схильні до нових мутацій. Звичайно, спонтанні мутації не залежить від статі. Відповідно, якщо у жінки-носія Х-хромосоми з мутацією гена F8 або F9 відбудеться спонтанна мутація в іншій Х-хромосомі, це призведе до класичної гемофілії. Тому жінки-носії гена гемофілії є групою ризику й потребують проведення відповідного скринінгу з визначенням рівня факторів згортання крові задля виключення або підтвердження наявності легкої гемофілії.
З точки зору лікарів акушерів-гінекологів, жінки, які є носіями гена гемофілії А і В, мають підвищений ризик розвитку кровотечі під час вагітності та пологів. Окрім того, ненароджена дитина може постраждати від наслідків порушення у системі гемостазу матері. Тому менеджмент жінок-носіїв гена гемофілії потребує залучення мультидисциплінарної команди фахівців, яка включатиме акушера-гінеколога, спеціаліста з питань вагітності високого ризику, лікаря-гематолога, дитячого гематолога, клінічного генетика та анестезіолога.
Оптимальний підхід до менеджменту жінок-носіїв гена гемофілії обов’язково має включати:
- генетичне консультування до вагітності;
- процедури пренатальної діагностики;
- план лікування для матері та дитини.
Незважаючи на дотримання всіх існуючих рекомендацій, жінки-носії гена гемофілії завжди матимуть ризик післяпологової кровотечі, тому розродження у них має відбуватися в медичних закладах третього (високоспеціалізованого) рівня надання медичної допомоги, у яких наявний досвід ведення пацієнток із гемофілією. Менеджмент цієї категорії пацієнток передбачає преконцепційну підготовку, антенатальне спостереження, розродження й післяпологовий нагляд, призначення контрацепції та подальше планування сім’ї.
Клінічний випадок 2
Пацієнтка Н., 23 років, була госпіталізована 27.06.2021 р. із приводу пологів. Пологи перші, своєчасні. Через 3 дні після пологів розвинулася маткова кровотеча. З приводу маткової кровотечі хворій було проведено операцію з вишкрібання матки. Кровотечу було зупинено. Важливо зазначити, що будь-які коагулопатії в анамнезі пацієнтки відсутні.
Дані лабораторних досліджень:
- гемоглобін – 81 г/л; еритроцити – 2,7×1012/л; тромбоцити – 298,0×109/л; ШОЕ – 47 мм/год;
- згортання крові: початок 7 хв 20 сек, кінець – за 10 хв кров не згорнулася;
- АЧТЧ – 85 сек;
- усі інші показники – у нормі.
Виписана з лікарні в задовільному стані, без ознак геморагічного синдрому.
Через тиждень – повторна маткова кровотеча. Госпіталізована до перинатального центру обласної лікарні з метою детальної діагностики та визначення причини кровотечі.
Дані лабораторних досліджень:
- АЧТЧ – 62 сек;
- протромбін – 84%;
- фібриноген – 1998 мг/л.
Хворій було призначено гемостатичну терапію окситоцином. Кровотечу було зупинено. Пацієнтка виписалася з лікарні в задовільному стані, без ознак геморагічного синдрому.
Через 3 тиж після виписки – повторна кровотеча, госпіталізована в гінекологічне відділення обласної лікарні.
Дані лабораторних досліджень:
- гемоглобін – 83-63 г/л; еритроцити – 2,22×1012/л; тромбоцити – 234,0-163,0×109/ л; ШОЕ – 40-60 мм/год;
- АЧТЧ – 62,1-85 сек;
- протромбін – 81-80%;
- фібриноген – не утворився;
- МНВ – 1,95.
Хворій було встановлено діагноз «Синдром дисемінованого внутрішньосудинного згортання» і призначено відповідну терапію, зокрема проведено операцію з приводу екстирпації матки та перев’язування внутрішніх клубових артерій. Після оперативного втручання кровотеча зменшилася, але повністю не припинилася.
На 6‑й день госпіталізації пацієнтка була проконсультована гематологом. До схеми лікування було додано рекомбінантний активований FVII (rFVIIa), що значно покращило клінічні симптоми й зупинило кровотечу. У хворої було запідозрено набуту коагулопатію. Для підтвердження цього діагнозу призначено лабораторну діагностику. Згідно з отриманими даними досліджень: активність FVIII <0,4%; інгібітори до FVIII – 38,9 БО/ мл. На підставі даних клінічної картини та лабораторних досліджень пацієнтці було встановлено діагноз «Набута гемофілія». Призначено метилпреднізолон у дозі 40 мг/добу + концентрат активованого протромбінового комплексу (activated prothrombin complex concentrate – aPCC) 3000 Од 2 рази на добу. На фоні отриманого лікування відзначалася позитивна клінічна й лабораторна динаміка.
Подальша стратегія менеджменту хворої включала консультацію у ДУ «Інститут патології крові та трансфузійної медицини НАМН України» (м. Львів), де їй було призначено лікування: метилпреднізолон 1-1,5 мг/кг/добу довготривало та препарати шунтуючої дії rFVIIa + aPCC 3000 Од 1 раз на добу.
Протягом останніх 2 міс проявів коагулопатії не спостерігалося. Пацієнтка продовжує отримувати метилпреднізолон 40 мг/добу та профілактичну дозу aPCC 3000 Од через день. Через 1,5 міс після початку лікування рівень інгібітора до FVIII становив 27,0 БО/мл, активність FVIII – 0,5%.
Станом на початок лютого 2022 року рівень інгібітора до FVIII становив 2,0 БО/ мл. Введення aPCC проводиться лише у випадку спонтанних кровотеч. Пацієнтка продовжує гормональну терапію за призначеною схемою.
З огляду на дані клінічного випадку, можна констатувати, що при веденні пацієнтки були допущені значні помилки. Зокрема, були невірно оцінені результати коагулограми, а саме такого показника, як АЧТЧ: його подовження потребує додаткового обстеження системи гемостазу, що не було проведено пацієнтці. Жінка була виписана зі змінами в коагулограмі, що призвело до рецидиву кровотечі. Крім того, такі кровотечі потребують обов’язкової консультації лікаря-гематолога.
Набута гемофілія – фатальний розлад гемостазу
За уявленням більшості лікарів, гемофілія є спадковим розладом системи гемостазу. Однак у клінічній практиці, окрім спадкової форми, відмічається набута гемофілія, а саме спонтанне аутоімунне захворювання – імунокоагулопатія. Його патогенез пов’язаний із синтезом специфічних аутоантитіл до одного із власних факторів згортання крові, найчастіше до FVIII – набута гемофілія А (НГА). НГА є рідкісним, загрозливим для життя й часто фатальним розладом згортання крові, який, на відміну від спадкової гемофілії А, може виникати й діагностуватися незалежно від статі й у будь-якому віці. Відомо, що захворюваність на НГА підвищується з віком, а пік захворюваності припадає на старшу вікову групу: за наявними даними, в осіб віком 65-84 роки частота НГА складає 9 випадків на 1 млн осіб на рік, натомість як в осіб віком >85 років – 15 випадків. При цьому у чоловічій популяції частота захворювання вища.
Ключовими чинниками розвитку НГА є злоякісні новоутворення (11,6%), аутоімунні захворювання (11,6%), вагітність (8,2%) та інші, менш поширені причини (інфекції, індукована ліками НГА, дерматологічні стани тощо), загальна частка яких складає 8,2%. І хоча сьогодні визначено стани, пов’язані з розвитком НГА, 52% випадків захворювання не мають відомої причини й є ідіопатичними.
Сучасні дані свідчать, що для НГА характерний двофазний віковий розподіл захворюваності: більш високий пік – серед жінок репродуктивного віку (20-40 років) та серед осіб віком >64 років; медіана віку – 33,9 року), наявність інгібітора до фактора згортання у яких асоціюється з вагітністю. Частота НГА, пов’язаної з вагітністю, становить 7-11% серед усіх випадків та <18% – серед жінок фертильного віку. Саме вагітні є переважаючою категорією пацієнтів із НГА молодого й середнього (працездатного) віку. НГА, пов’язана з вагітністю, зазвичай відмічається не у період гестації, а після пологів і може виникати вже на 3‑й день, а загалом – протягом 1-6 міс після пологів.
Враховуючи клінічну картину та ризик для пацієнта, критичним є час до встановлення діагнозу НГА та призначення відповідного лікування. Сьогодні клініцисти мають у розпорядженні значну кількість гайдлайнів щодо діагностики й лікування НГА. Зокрема, останній гайдлайн датований 2020 роком (Tiede A. et al., 2020). Розробки у цій галузі продовжуються, й Україна не є винятком: наразі триває розробка Національного протоколу з діагностики й лікування НГА, а також Національного реєстру НГА. Зараз вітчизняні лікарі користуються міжнародними стандартами. За даними існуючих на сьогодні у світі реєстрів, зокрема європейського, у 38,2% випадків НГА діагноз встановлюється у 1‑й день, у 26,5% – приблизно на 4‑й день, у 35,3% – у середньому через 20 днів від початку кровотечі. Головна причина затримки – недостатність лабораторної бази й лікарської обізнаності щодо цієї патології. Діагностичні затримки часто відтерміновують старт гемостатичної терапії НГА, що, відповідно, негативно позначається на перебігу захворювання й підвищує ризик смерті пацієнтів.
Із проблемою НГА можуть стикатися у рутинній практиці лікарі будь-якої спеціальності – хірурги, неврологи, стоматологи та ін. Тому рівень поінформованості щодо цієї патології серед лікарів має підвищуватися. Імовірність діагнозу НГА слід розглядати у пацієнтів із аномальною кровотечею та подовженим АЧТЧ за відсутності особистого або сімейного анамнезу розладів згортання крові.
Враховуючи, що НГА є досить рідкісним розладом системи гемостазу, який виникає несподівано, пацієнтів можуть спостерігати лікарі різних спеціальностей. Тому необхідний спрощений діагностичний алгоритм для допомоги лікарям, які, можливо, не мають прямого досвіду ведення пацієнтів із НГA (рис. 1).
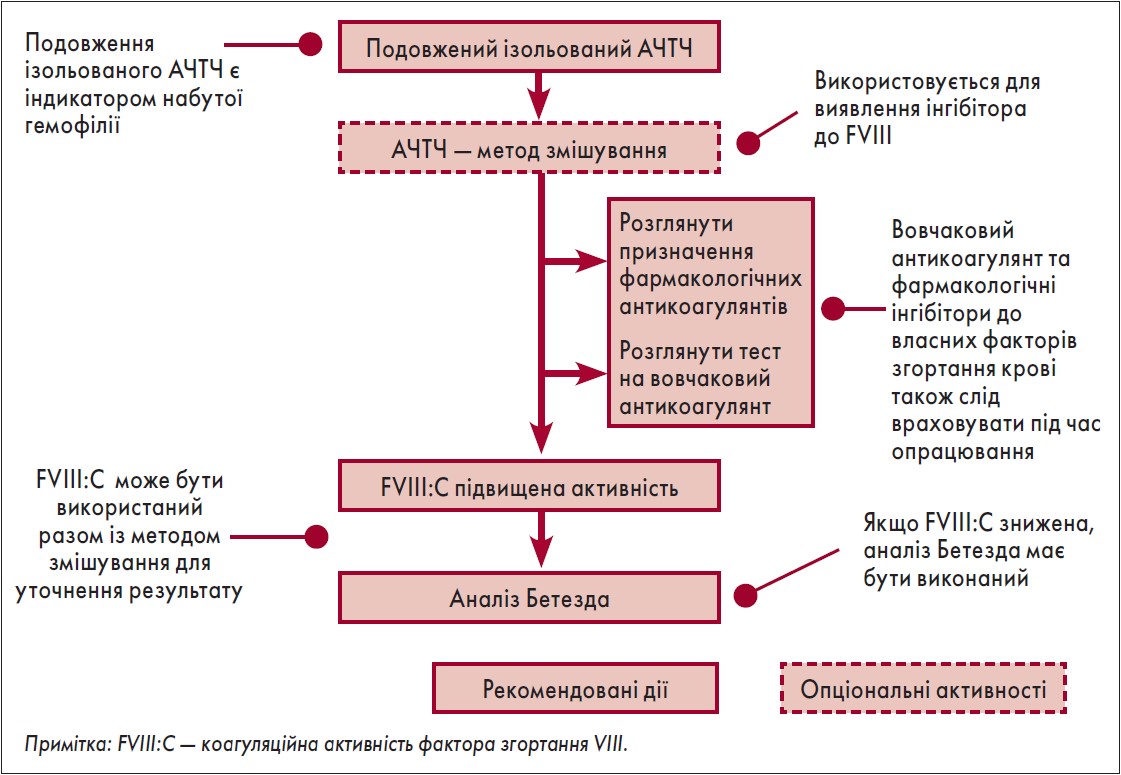
Рис. 1. Алгоритм діагностики НГА
У світлі сучасних міжнародних рекомендацій лікуванням НГА повинні керувати спеціалісти з гемофілії, незалежно від клінічної картини захворювання. Слід пам’ятати, що гемостатичні засоби не завжди мають передбачувану ефективність, а також, поряд з імуносупресивною терапією, супроводжуються значними ризиками для пацієнта. Фундаментальними аспектами терапевтичної тактики менеджменту НГА є зупинка кровотечі, ерадикація інгібітора до фактора згортання та лікування супутніх захворювань і станів. Відповідно до сучасних рекомендацій, стратегії зупинки кровотечі у хворих із НГА передбачають застосування шунтуючих препаратів: aPCC, rFVIIа або рекомбінантного свинячого FVIII. Для ерадикації інгібітора до факторів згортання крові показана імуносупресивна терапія із застосуванням глюкокортикостероїдів як препаратів першої лінії. За неефективності лікування першої лінії рекомендоване застосування цитостатиків: циклофосфаміду, ритуксимабу (рис. 2).
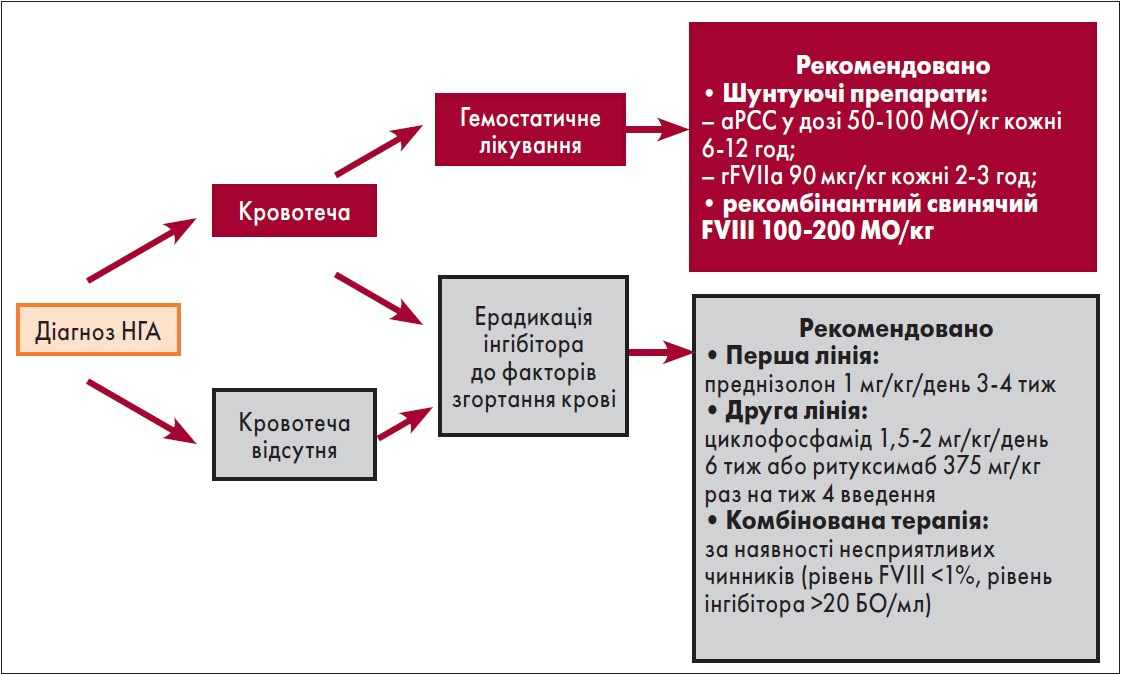
Рис. 2. Алгоритм лікування НГА
Отже, набута гемофілія, спричинена розвитком інгібіторів до власних факторів згортання крові, є рідкісною й тяжкою патологією, що потребує залучення до лікувально-діагностичного процесу не лише лікаря акушера-гінеколога, а й гематолога. Такий мультидисциплінарний підхід дозволяє забезпечити повноцінне обстеження й специфічне лікування пацієнток із АМК та АК. Відповідно до сучасних гайдлайнів, гостру кровотечу необхідно якнайшвидше купірувати: для цього доцільно застосовувати препарати із шунтуючим механізмом дії, у тому числі aPCC, показані як терапія першої лінії.
Підготувала Анна Хиць
За підтримки ТОВ «Такеда Україна»,
VV-MEDMAT-81578.
