


1 листопада, 2020
Хвороба Гоше: must know для педіатрів

![]() Орфанні (рідкісні) захворювання – це велика гетерогенна група, що об’єднує близько 6-8 тис. нозологій, для яких характерний тяжкий хронічний прогресуючий перебіг із формуванням дегенеративних змін в організмі, які з часом призводять до інвалідизації пацієнта. Для деяких захворювань існує високоефективне специфічне лікування, яке не лише зменшує або усуває симптоми, але й суттєво покращує якість життя хворих та дає шанс на нормальний розвиток, що є особливо цінним у дитячому віці. Тому лікарям первинної ланки медичної допомоги вкрай важливо бути настороженими щодо рідкісних захворювань, адже від них залежить своєчасність встановлення правильного діагнозу та призначення специфічної терапії.
Орфанні (рідкісні) захворювання – це велика гетерогенна група, що об’єднує близько 6-8 тис. нозологій, для яких характерний тяжкий хронічний прогресуючий перебіг із формуванням дегенеративних змін в організмі, які з часом призводять до інвалідизації пацієнта. Для деяких захворювань існує високоефективне специфічне лікування, яке не лише зменшує або усуває симптоми, але й суттєво покращує якість життя хворих та дає шанс на нормальний розвиток, що є особливо цінним у дитячому віці. Тому лікарям первинної ланки медичної допомоги вкрай важливо бути настороженими щодо рідкісних захворювань, адже від них залежить своєчасність встановлення правильного діагнозу та призначення специфічної терапії.
У рамках II Міжнародного конгресу PRIME Pediatrics 2020, який цього року відбувся в режимі онлайн, кандидат медичних наук Анна Михайлівна Гільфанова (Національна медична академія післядипломної освіти імені П.Л. Шупика) представила доповідь «Медичний супровід пацієнтів із хворобою Гоше: must know для педіатра».
– Для лікування, профілактики та діагностики рідкісних захворювань використовуються так звані орфанні препарати (наразі нараховується близько 200-300 подібних лікарських засобів), які зазвичай є дороговартісними. Тому для забезпечення доступу орфанних пацієнтів до специфічного лікування необхідна цілеспрямована державна політика. У 2014 р. був прийнятий Закон України «Про внесення змін до Основ законодавства України про охорону здоров’я щодо забезпечення профілактики та лікування рідкісних (орфанних) захворювань», який гарантує безперервне та безоплатне забезпечення лікарськими засобами громадян із рідкісними хворобами. Тому педіатр як лікар первинної ланки медичної допомоги несе відповідальність за своєчасне припущення наявності орфанного захворювання, щоб дитина могла отримати необхідне лікування, яке забезпечить їй подальше повноцінне життя.
У структурі поширеності рідкісних захворювань 2-ге місце посідає група спадкових порушень обміну речовин, або метаболічні захворювання, серед яких найрозповсюдженішою патологією є хвороба Гоше (ХГ): у середньому 1 випадок на 40 тис. населення. ХГ (глюкоцереброзидний ліпідоз, глюкоцереброзидоз) – це лізосомальне захворювання з аутосомно-рецесивним типом успадкування. Патологія виникає при наявності двох мутацій алелі гена GBA (гомозиготний стан або гетерозиготний компаунд), що призводить до зниження активності глюкоцереброзидази. Як наслідок, неутилізовані ліпіди накопичуються в лізосомах макрофагів кісткового мозку, селезінки, печінки, лімфатичних вузлів, кісток, нейронів головного мозку, легенів, що зумовлює мультисистемний прояв хвороби з утворенням так званих клітин накопичення (клітин Гоше). Хронічна активація макрофагальної системи призводить до аутокринної стимуляції моноцитопоезу та збільшення абсолютної кількості макрофагів, що в свою чергу супроводжується порушенням їхньої регуляторної функції, тобто у таких пацієнтів уражається вроджена ланка імунітету.
Зниження активності ферменту є дуже варіабельним, що впливає на клінічний спектр ХГ. Виділяють 3 основні типи ХГ: ненейропатичний, гострий нейропатичний, підгострий нейропатичний. Найрозповсюдженішим варіантом є тип 1, який слід запідозрити при наявності наступних клінічних ознак:
- затримка фізичного та статевого розвитку;
- слабкість, підвищена втомлюваність (астенія);
- часті респіраторні інфекції;
- ознаки геморагічного синдрому (підшкірні гематоми, підвищена кровоточивість слизових оболонок, тривалі кровотечі при малих оперативних втручаннях);
- біль у кістках та суглобах, часті переломи кісток;
- неврологічна симптоматика (окорухова апраксія або збіжна косоокість, атаксія, зниження інтелекту, порушення чутливості);
- сімейний анамнез (наявність спленектомії або вище наведених симптомів у рідних братів чи сестер);
- виявлені при фізикальному обстеженні хворого ознаки геморагічного синдрому, гепатоспленомегалії, лімфаденопатії, гіперпігментації шкіри в області ліктьових та колінних суглобів.
Серед перелічених ознак у пацієнтів із ХГ найчастіше виявляють гепатомегалію (87%), спленомегалію (95%), анемію (40%), тромбоцитопенію (50%), колби Ерленмеєра (49%), інфільтрацію кісткового мозку (39%), біль у кістках (27%), остеопенію (20%), затримку росту (<5 перцентиля – 28% і в межах 5-25 перцентиля – 28%).
Спленомегалія є найпоширенішим проявом ХГ. У деяких випадках селезінка може досягати гігантських розмірів, що візуально проявляється значним збільшенням об’єму живота у дитини. Разом із гіперспленізмом (надмірна маса та розміри селезінки), що призводить до цитопенії, у таких пацієнтів виникає функціональний гіпоспленізм, який впливає на імунітет та підвищує схильність до розвитку бактеріальних інфекцій.
Кістки найчастіше уражуються при ХГ типу 1. Патогномічним симптомом захворювання є колби Ерленмеєра – колбовидна деформація дистальних відділів стегнових кісток. До локальних уражень кісткової системи належить інфаркт кістки з наступним формуванням склерозу та патологічних переломів. Генералізоване ураження кісток проявляється остеопорозом та остеопенією, переломами хребців, які можуть призвести до компресії спинного мозку. Локальний тромбоз судини, що живить голівку епіфізу стегнової кістки, викликає асептичний некроз. У таких випадках пацієнт потребує оперативного лікування з протезування суглобу.
Для ХГ також характерні кісткові кризи, які супроводжуються виснажливим болем у кістках протягом кількох днів чи тижнів, гіперемією шкіри над ураженими суглобами, різким зниженням рухової активності, значним розширенням кістково-мозкового каналу. Така клінічна картина дуже нагадує остеомієліт, проте у випадку ХГ відсутня лихоманка та зміни гемограми, характерні для запалення. При підозрі на хворобу Гоше для встановлення остаточного діагнозу слід провести такі дослідження:
- вимірювання активності глюкоцереброзидази в сухих плямах крові як скринінг-метод, який дозволяє зменшити строки діагностики ХГ;
- вимірювання активності глюкоцереброзидази у лейкоцитах крові, що є високоточним методом, який дозволяє встановити діагноз ХГ;
- вимірювання активності хітотриозидази є допоміжним тестом при діагностиці ХГ, оскільки значна частка населення є носіями алелей, що призводять до низької активності цього ферменту. Крім того, активність хітотриозидази є важливим маркером успішності лікування ХГ;
- молекулярно-генетична діагностика з метою виявлення мутацій у гені глюкоцереброзидази.
Гострий нейропатичний (інфантильний) тип ХГ дебютує у віці 3-5 міс. Початковими проявами є м’язова гіпотонія, втрата цікавості дитини до оточення, затримка і регрес психомоторного розвитку. Про прогресування цього типу ХГ свідчить поява спастики, окорухових порушень, ларингоспазму, проблем із вигодовуванням (через бульбарні порушення, тризм), спленомегалії, тоніко-клонічних судом, синдрому «вишневої кісточки» (не постійна ознака). При магнітно-резонансній томографії (МРТ) у таких хворих виявляють ознаки церебральної атрофії та дурального потовщення, а при аутопсії – нейронофагію. На сьогодні не існує ефективних методів лікування ХГ типу 2, і хворі діти помирають до 2-річного віку.
ХГ типу 3 поділяється на ювенільний та шведський підтипи, які характеризуються широким спектром неврологічних проявів. При ювенільному підтипі дебют захворювання припадає на перші декади життя і супроводжується гепатоспленомегалією, що повільно прогресує, менш тяжкими, ніж при ХГ типу 2, неврологічними проявами (горизонтальна офтальмоплегія, міоклонії, генералізовані тоніко-клонічні судоми, зниження інтелекту, екстрапірамідна ригідність, мозочкові порушення, розлади мовлення та письма, поведінкові розлади, епізоди психозу, втрата слуху). При шведському підтипі дебют хвороби припадає на 1 рік життя, а клінічними ознаками є швидко прогресуюча гепатоспленомегалія, затримка психічного та мовленнєвого розвитку, атаксія, спастичний тетрапарез, який повільно прогресує до 3 років.
Окремо виділяють фетальну форму ХГ, яка становить 5% від усіх випадків захворювання. Хвороба маніфестує внутрішньоутробно та проявляється зменшенням або відсутністю рухів плода, фетальною та плацентарною анасаркою, гепатоспленомегалією, іхтіозом, артрогрипозом, лицевим дизморфізмом, фетальною тромбоцитопенією. У таких випадках смерть настає внутрішньоутробно або протягом 3 міс після народження.
ХГ слід диференціювати зі злоякісними гематологічними захворюваннями, тромбоцитопенічною пурпурою, хронічними захворюваннями печінки, остеомієлітом, туберкульозом кісток, гемофілією, анемією, затримкою розвитку дитини тощо. У разі підозри ХГ при фізикальному обстеженні пацієнту необхідно провести наступні дослідження: розгорнутий аналіз крові (для визначення рівня гемоглобіну та кількості тромбоцитів), біохімічний аналіз крові (для визначення рівня ліпідів, холестерину, протромбінового часу, рівня імуноглобулінів, АПФ, ТРКФ, рівня феритину), УЗД або МРТ селезінки та печінки, рентгенографію або МРТ поперекового відділу хребта та стегнових кісток. Ці обстеження можуть підтвердити зміни, характерні для ХГ.
Наступним етапом діагностики є визначення активності ферменту β-D-глюкозидази в лейкоцитах периферійної крові (культура клітин), у сухих плямах крові та культурі фібробластів (біоптат шкіри). Слід враховувати те, що ступінь зниження активності ферменту не корелює з тяжкістю клінічних проявів захворювання. Додатковим біохімічним маркером ХГ є підвищення активності хітотриозидази – ферменту, який синтезується клітинами Гоше. На тлі лікування активність ферменту знижується, тому цей показник використовується для оцінки ефективності терапії. Молекулярно-генетична діагностика проводиться з метою виявлення мутацій у гені глюкоцереброзидази.
Отже, педіатр та сімейний лікар є першими спеціалістами, які контактують із пацієнтами з ХГ. Тому вкрай важливо вчасно припустити наявність патології, призначити базові лабораторні та інструментальні дослідження (табл.), проконсультуватися з лікарем-гематологом та скерувати пацієнта до лікаря-генетика, який визначить наступні дії та склад мультидисциплінарної команди для тривалого спостереження за хворим.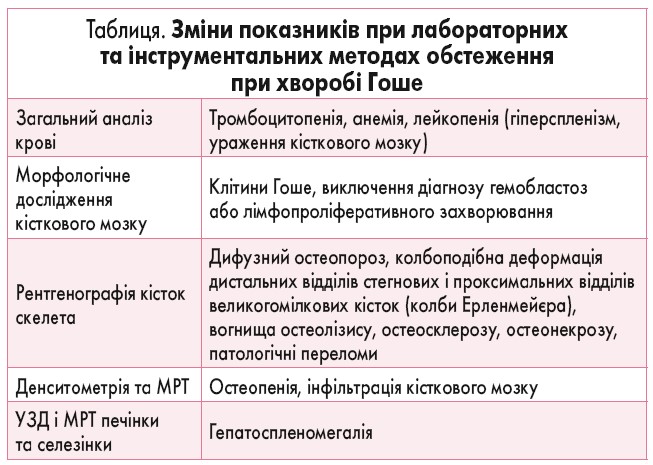
Основною складністю своєчасного встановлення діагнозу ХГ є низька настороженість лікарів щодо цієї нозології. Період від моменту виникнення перших симптомів до встановлення діагнозу може сягати 4-10 років. Кожен пацієнт протягом цього часу в середньому звертається до 8 різних фахівців.
Ефективним та безпечним методом лікування ХГ (окрім нейропатичного типу ХГ) є довічна ферментозамісна терапія (ФЗТ) рекомбінантною глюкоцереброзидазою. ФЗТ усуває основні симптоми захворювання та покращує якість життя хворого без виражених побічних ефектів. Частота введення препарату – 1 раз на 14 днів в/в крапельно протягом 1,5-2 год. Корекція дози ФЗТ здійснюється досвідченим лікарем із огляду на досягнення та утримання терапевтичних цілей для кожного пацієнта індивідуально.
Сьогодні в Україні зареєстровані такі препарати для ФЗТ:
- таліглюцераза (Елелісо, Pfizer) – рекомбінантний аналог лізосомальної глюкоцереброзидази людини, виробляється за технологією рекомбінантної ДНК із використанням культури рослинних клітин (моркви);
- велаглюцераза альфа – людська лінія фібробластів; амінокислотна послідовність у препараті ідентична ендогенній глюкоцереброзидазі;
- іміглюцераза – клітинна лінія, отримана з яєчників китайських хом’яків.
Вибір препарату для ФЗТ для кожного пацієнта здійснюється колегіально.
Крім основного лікування, хворому на ХГ призначається симптоматичне лікування (при остеопорозі – дієта, збагачена кальцієм, вітаміном D, при кістковому кризі – аналгетики, при бактеріальній інфекції – антибактеріальну терапію, ортопедичне лікування, при неврологічній симптоматиці – ноотропи, протиепілептичні препарати, міорелаксанти тощо). Також при веденні пацієнта з ХГ важливе значення має профілактика сепсису при функціональному гіпоспленізмі, основним напрямком якої є вакцинація проти пневмококової, менінгококової та Hib-інфекції.
Крім того, пацієнти з рідкісними захворюваннями потребують інформаційної, соціальної, психологічної та юридичної підтримки, яку надають громадські та пацієнтські організації: ГО «Орфанні захворювання України», ГО «Всеукраїнське об’єднання інвалідів – хворих на хворобу Гоше».
Підготувала Ілона Цюпа
Статтю надруковано за підтримки представництва компанії «Пфайзер Eкспорт Бі. Ві.» в Україні.
PP-ELE-UKR-0023
Тематичний номер «Педіатрія» №4 (55) 2020 р.
