


11 лютого, 2023
Гіпоспадія як прояв порушення статевого диференціювання Клінічні випадки
За матеріалами конференції
 3-4 вересня минулого року в Києві відбулася щорічна науково-практична конференція «Осіння зустріч Європейської спільноти андрологічної урології», яку було присвячено обговоренню важливих питань чоловічого репродуктивного здоров’я. У рамках заходу з доповіддю «Гіпоспадія як прояв порушення статевого диференціювання. Клінічні випадки» виступила завідувачка відділу дитячої ендокринології Українського науково-практичного центру ендокринної хірургії, трансплантації ендокринних органів і тканин МОЗ України, заслужений лікар України, доктор медичних наук, професор Наталія Борисівна Зелінська.
3-4 вересня минулого року в Києві відбулася щорічна науково-практична конференція «Осіння зустріч Європейської спільноти андрологічної урології», яку було присвячено обговоренню важливих питань чоловічого репродуктивного здоров’я. У рамках заходу з доповіддю «Гіпоспадія як прояв порушення статевого диференціювання. Клінічні випадки» виступила завідувачка відділу дитячої ендокринології Українського науково-практичного центру ендокринної хірургії, трансплантації ендокринних органів і тканин МОЗ України, заслужений лікар України, доктор медичних наук, професор Наталія Борисівна Зелінська.
Ключові слова: гіпоспадія, мутації, порушення статевого диференціювання.
Доповідач нагадала, що формування статі ембріона бере свій початок від мезодерми (рисунок). Під впливом генів WT1 (Wilm’s tumor 1) та SF-1 (steroidogenic factor 1) формується первинний гонадний тяж. Надалі з нього виникає недиференційована гонада, з якої, залежно від наявності певного гена, формуються яєчники або яєчка. До 8-го тижня вагітності усі первинні статеві структури плода ідентичні, після чого вже під впливом тестостерону та дигідротестостерону формується чоловічий фенотип. Без впливу андрогенів (їх відсутності чи недостатності) розвивається жіночий або проміжний фенотип.
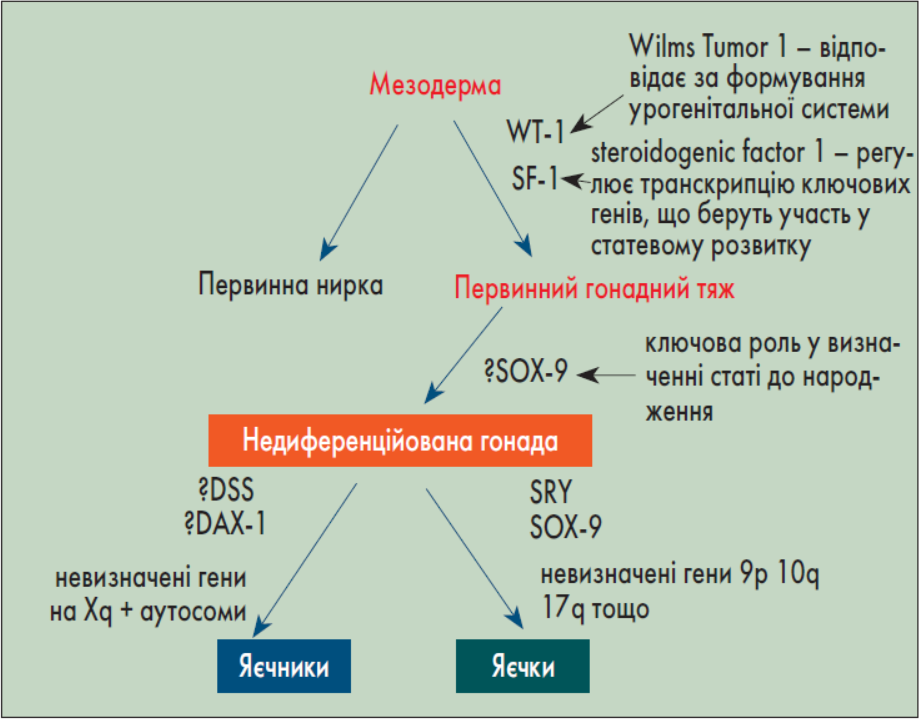
Рис. Формування статі
Диференціювання чоловічої статі відбувається за певною хронологією. На 7-му тижні вагітності клітини Сертолі продукують антимюллерів гормон (АМГ), який призводить до регресії мюллерових проток. Протягом 8-го тижня клітини Лейдіга виробляють тестостерон, що дозволяє формуватися вольфовим протокам. З 8-го по 13-й тиждень тестостерон під впливом 5-альфа-редуктази перетворюється на дигідротестостерон, який діє на рецептори андрогенів, що сприяє розвиненню зовнішніх чоловічих геніталій. У разі відсутності андрогенів, або їх дефіциту, або нечутливості до них у дитини формується жіночий чи проміжний фенотип.
Гіпоспадія – це вроджений дефект, який виникає під час ембріонального розвитку уретри на 8-13-му тижні вагітності. Дана патологія відзначається із частотою 1:300 новонароджених дітей чоловічої статі й частіше зустрічається у однояйцевих близнюків. Також високий відсоток виникнення гіпоспадії спостерігається при сімейному успадкуванні, у тому числі в осіб 1-го, 2-го та 3-го ступенів спорідненості.
За сучасною класифікацією (2003) гіпоспадію розрізняють на передню (головчаста, вінечна, передньо-стовбурова), дистальну (середньо-стовбурова) й проксимальну (задня, задньо-стовбурова, стовбурово-калиткова, калиткова, промежинна).
До найбільш частих аномалій, які асоціюються з гіпоспадією, відносяться:
- крипторхізм;
- пахова грижа;
- гідроцеле;
- відкритий піхвовий відросток;
- аномалії верхніх сечових шляхів;
- простатична маточка;
- неповна регресія мюллерових проток;
- вроджена дисфункція кори надниркових залоз (ВДКНЗ, адреногенітальний синдром);
- змішаний гонадальний дисгенез.
Близько 30% дітей із гіпоспадією мають порушення статевого розвитку (disorders of sexual development, DSD). M. Kaefer et al. (1999) виявили DSD майже у половини дітей із двостороннім крипторхізмом, що пов’язаний із гіпоспадією. Тому при поєднанні гіпоспадії будь-якого ступеня із крипторхізмом, мікропенією, неоднозначними статевими органами або деформацією калитки необхідне повне генетичне та ендокринологічне обстеження одразу після народження дитини з метою виключення DSD.
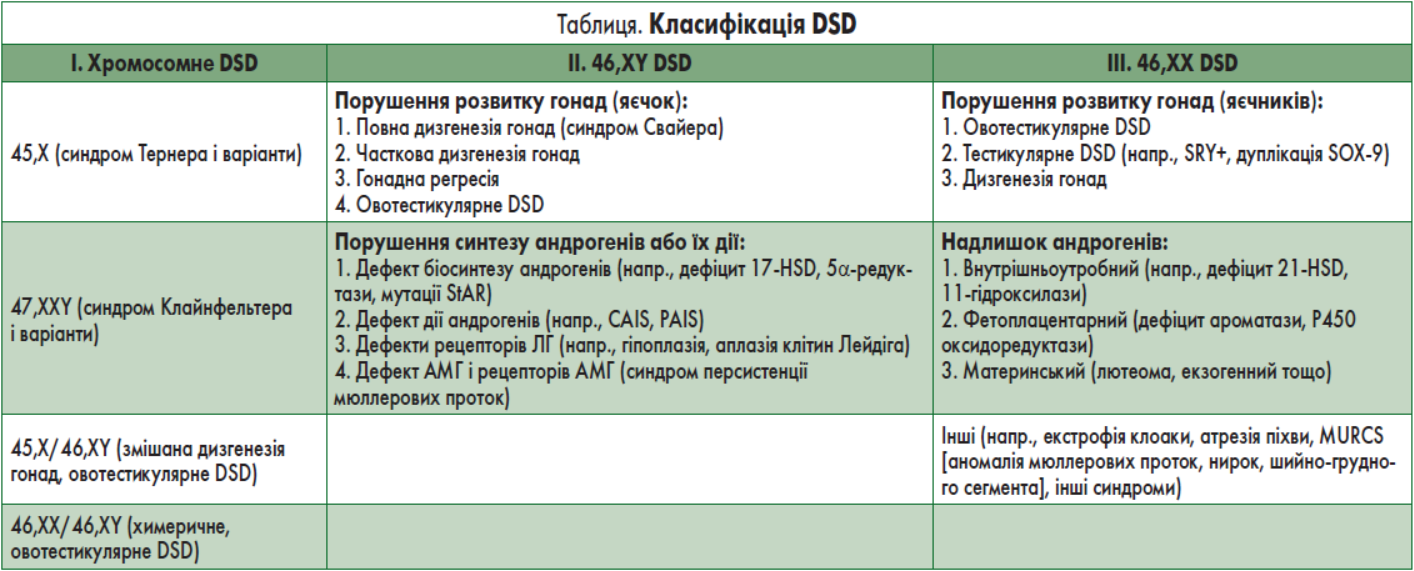
В продовжені доповіді Наталія Борисівна продемонструвала класифікацію DSD, якою користуються у всьому світі (таблиця).
Діагностику пацієнтів із проксимальною гіпоспадією слід починати з оцінки сімейного анамнезу та каріотипування. За допомогою лабораторних методів можна визначити рівень гормонів – фолікулостимулюючого (ФСГ), лютеїнізуючого (ЛГ), тестостерону, естрадіолу, інгібіну В, антимюллерового гормона (АМГ). Серед інструментальних методів призначають ультразвукове дослідження (УЗД) гонад, магнітно-резонансну томографію (МРТ) або ретроградну генітографію, цистоскопію та мікційну цистоуретрографію. За необхідності діагностику можна доповнити гістологічним дослідженням гонад та молекулярно-генетичним дослідженням. Доповідач зауважила, що діти з ізольованою передньою та дистальною гіпоспадією не потребують додаткового поглибленого обстеження.
Із 2000 року на базі медико-генетичного центру НДСЛ «Охматдит» започатковано реєстр дітей із DSD. У 2016 році його було об’єднано з реєстром хворих Українського науково-практичного центру ендокринної хірургії, трансплантації ендокринних органів та тканин МОЗ України. Критеріями включення до даного реєстру є неправильна або невизначена будова зовнішніх статевих органів та/або невідповідність гонадної статі хромосомній.
Професор Н.Б. Зелінська докладно висвітлила найпоширеніші мутації генів, що викликають гіпоспадію.
Мутації в гені андрогенового рецептора (AR). Нечутливість до дії андрогенів може бути повною (CAIS) або частковою (РAIS) і має X-зчеплене успадкування. На відміну від інших форм 46,XY DSD у пацієнтів є сліпий піхвовий мішок та відсутні мюллерові структури (наявні яєчка з нормальною продукцією АМГ). Залежно від ступеня нечутливості до андрогенів клінічні прояви можуть коливатись від нормального чоловічого фенотипу з безпліддям до наявності геніталій невизначеної будови й аж до формування повністю жіночого фенотипу з нормальними молочними залозами, але з одно- чи двобічними «паховими грижами» (що насправді є яєчками), а приводом до звернення до лікаря є первинна аменорея в пубертаті.
Спікер навела шість випадків мутації в гені AR у реєстрі дітей із DSD, з яких у п’яти хворих спостерігався жіночий фенотип (CAIS), і їх було зареєстровано у жіночій статі. В одного хворого з PAIS, якого було зареєстровано у чоловічій статі, відзначалися проксимальна гіпоспадія, гіпоплазія статевого члена та лівобічна пахова грижа. Лікування препаратами тестостерону в таких пацієнтів є недоцільним через відсутність терапевтичного ефекту.
Мутація в гені NR5A1 (або SF1, стероїдний фактор 1). Даний ген експресується в урогенітальному тракті, гіпоталамусі, передній долі гіпофіза та наднирникових залозах. У хворих 46,XY із мутаціями в гені NR5A1 можливий широкий діапазон фенотипів – від чоловічого безпліддя, легкої гіповірилізації з гіпоспадією та/або крипторхізмом до тяжкої гіповірилізації та геніталій невизначеного типу. Гонадний фенотип може бути від нормального чоловічого до дизгенетичного та анорхії.
Було представлено клінічні випадки чотирьох дітей із такою мутацією. Двоє хлопчиків-близнюків із помірною гіповірилізацією, двобічним крипторхізмом, проксимальною гіпоспадією та мікропенією мали сімейний анамнез DSD у батька й дідуся; народження хлопчиків-близнюків стало можливим лише завдяки екстракорпоральному заплідненню. Двоє інших дітей бути зареєстровані у жіночій статі. Першій дівчинці із кліторомегалією та урогенітальним синусом у віці 1,5 міс встановлено хибний діагноз ВДКНЗ, призначено глюкокортикоїди, які було відмінено у 4 роки після уточнення діагнозу. У другої дівчинки при народженні зовнішні статеві органи (ЗСО) проміжного типу, урогенітальний синус, кліторомегалія, гонади у пахових каналах.
Мутації в генах АМГ та рецепторах АМГ типу 2 (AMHR2). Синдром персистенції мюллерових проток (СПМП) виникає внаслідок мутації в гені АМГ. Це рідкісний синдром, що характеризується відсутністю регресії дериватів мюллерових проток в осіб чоловічої статі. Існує два типи СПМП: перший тип пов’язаний із мутацією самого гена АМГ, другий – із мутацією в гені AMHR2.
У якості прикладу СПМП доповідач представила клінічний випадок хлопчика, який народився з гіпоспадією та відсутністю яєчок у калитці. У віці 1 року йому було виконано ревізію пахового каналу з приводу «пахової грижі». При УЗД малого таза було виявлено матку й гонади у черевній порожнині, що зумовило дослідження каріотипу, який виявився нормальним чоловічим (46,ХY). При уточненні сімейного анамнезу з’ясовано, що у старшого брата при народженні також мав місце паховий крипторхізм і гіпоспадія, а при цілеспрямованому обстеженні у віці 15 років у пацієнта виявлено матку. Обом хлопчикам було проведене повне екзомне секвенування й виявлено мутації в гені AMHR2, два різні варіанти якої були успадковані від матері та батька.
Мутації в гені HSD17B3 призводять до дефіциту ферменту 17β-HSD типу 3, що бере участь у стероїдогенезі, перетворюючи андростендіон на більш активний тестостерон. Дефіцит 17β-HSD-3 – найпоширеніший дефект біосинтезу тестостерону в осіб із 46,XY DSD. У таких пацієнтів ЗСО – від переважно жіночих або неоднозначних до переважно чоловічих із мікропенією та гіпоспадією. Жіночі зміни виникають частіше й проявляються зрощенням статевих губ, піхвою зі сліпим кінцем, можливою кліторомегалією. За невчасної діагностики у підлітковому віці виникає значна вірилізація та первинна аменорея. Тому кожна дівчинка з паховою грижею, незначною кліторомегалією або урогенітальним синусом має підозру на дефіцит 17β-HSD типу 3.
До реєстру увійшло двоє таких хворих жіночої статі. Першій дівчинці у 8-місячному віці було проведено хірургічну пластику двобічної пахової грижі без проведення дообстеження. У 10 років у пацієнтки стався рецидив даної патології: методом УЗД виявлено відсутність матки та гонади у паховому каналі. В іншої дівчинки спостерігалися зовнішні статеві органи проміжного типу (скротолабіальні складки, промежинна гіпоспадія).
Мутації в гені MYRF (Myelin regulatory factor) асоціюються із вродженою діафрагмальною грижею, енцефалопатією, серцевими та урогенітальними аномаліями. Відомо, що MYRF впливає на мієлінізацію у центральній нервовій системі.
Було виявлено двоє хворих із мутацією в гені MYRF. Хлопчик із зовнішніми статевими органами проміжного типу – промежинна гіпоспадія, двобічний крипторхізм. За даними біопсії яєчок виявлено, що їх тканина з вираженим склерозом строми та атрофією канальців. Інший приклад – дівчинка, яка звернулась до ендокринолога у 14 років із приводу первинної аменореї та відсутності вторинних статевих ознак. При обстеженні виявлено первинний гіпогонадизм та гірсутизм. За даними УЗД матка та гонади розташовані у черевній порожнині.
Мутація в гені DHX37 (DEAH-box helicase 37) є частою причиною несиндромної 46,XY дисгенезії гонад і синдрому тестикулярної регресії. Даний ген експресується у соматичних клітинах яєчка та відіграє ключову роль у ранній детермінації та підтримці тканин яєчок під час ранньої фази їх розвитку.
У якості клінічного випадку було представлено дівчинку з паховими грижами, у якої при подальшому обстеженні виявлено первинний гіпогонадизм, урогенітальний синус та гонади у черевній порожнині. У хлопчика із синдромом тестикулярної регресії було виявлено мутацію в гені DHX37, проте без явної клінічної значущості.
Мутація в гені KAL1 (Kallmann 1) спричиняє синдром Каллмана з Х-зчепленим успадкуванням. В основі даної патології лежить порушення імпульсної секреції гонадоліберину в гіпоталамусі. Клінічно мутація в гені KAL1 проявляється гіпогонадотропним гіпогонадизмом з аносмією або гіпоосмією.
З даною генною мутацією було виявлено трьох хворих. Перший випадок – дитина, що при народженні мала зовнішні статеві органи проміжного типу, скротолабіальні складки, урогенітальний синус та промежинну гіпоспадію. За даними УЗД права гонада була виявлена у скротолабіальній складці, ліва – у паховому каналі. До 4-місячного віку дитина не мала визначеного статевого статусу, однак за рішенням консиліуму віднесена до чоловічої статі. Інші два випадки – двоюрідні брати із двобічним паховим крипторхізмом та голівчастою гіпоспадією, що в ранньому дитинстві прооперовані урологом. Перші звернення до ендокринолога відмічені у 12 та 13 років відповідно із причини відсутності належного пубертату.
Професор Н.Б. Зелінська навела нещодавній клінічний випадок пацієнта 13,5 років, який вперше звернувся до ендокринолога з приводу гінекомастії. З анамнезу було відомо, що при народженні у хворого діагностовано промежинну гіпоспадію (двічі оперований), крипторхізм (тричі орхіпексія) без чіткого виявлення лівого яєчка. У віці 3 років хлопчику було проведено каріотипування (каріотип 46,ХY). За даними МРТ передміхурова залоза та ліве яєчко відсутні. При гормональному обстеженні виявлено підвищені рівні у крові ФСГ, ЛГ, вільного та загального тестостерону. Пацієнт був направлений на генетичне обстеження, яке виявило мутації в гені AR , що підтвердило попередній діагноз синдрому нечутливості до андрогенів (PAIS).
Отже, генні мутації та їх наслідки є серйозною не лише клінічною, а й психологічною та соціальною проблемою.Ці фізіологічні порушення потребують своєчасної уваги лікарів із застосуванням сучасної діагностики. З огляду на це дітям із дистальною та проксимальною гіпоспадією або іншими урогенітальними аномаліями необхідно при першому ж зверненні проводити повноцінне генетичне та ендокринологічне обстеження з метою виключення DSD
Підготувала Оксана Габрук
