


10 березня, 2021
Сучасні виклики та рідкісна патологія в практичній ендокринології
Науково-освітній проект «Школа ендокринолога»
На черговій онлайн-конференції в рамках науково-освітнього проекту «Школа ендокринолога», яка відбулася наприкінці листопада 2020 р., слухачі мали можливість прослухати багато цікавих доповідей провідних фахівців України. Темами виступів стали актуальні проблеми та основні сучасні тенденції лікування та діагностики в клінічній ендокринології.
Проблемним питанням цукрового діабету (ЦД) за наявності коронавірусного захворювання присвятив свій виступ науковий керівник науково-освітнього проекту «Школа ендокринолога» – директор ДУ «Інститут ендокринології і обміну речовин ім. В.П. Комісаренка НАМН України» (м. Київ), академік НАМН України, член-кореспондент НАН України, завідувач кафедри ендокринології Національної медичної академії післядипломної освіти ім. П.Л. Шупика МОЗ України (м. Київ), доктор медичних наук, професор Микола Дмитрович Тронько.
30 січня 2020 року Всесвітня організація охорони здоров’я (ВООЗ) оголосила про спалах COVID‑19 у світі. У різних странах на всіх континентах виявлено та діагностовано багато людей із цією нозологією. З березня 2020 року, унаслідок збільшення кількості хворих у світі, епідемію трансформували в пандемію.
На 25 листопада 2020 року на COVID‑19 захворіли 59 533 146 осіб, із них одужало – 41 173 110, 1 402 312 – померло. Найбільша захворюваність спостерігається в США (12 777 371 особа), де щоденно реєструють більш як 100 тис нових інфікованих. Щодо європейських країн, таких як Італія, Іспанія, Франція, Німеччина, Велика Британія, там зареєстровано 1,5-2 млн хворих. У Росії – 2 344 000.
В Україні станом на цей же період обстежено на COVID‑19 загалом 4 279 025 осіб, у 647 976 з них виявлено інфекцію, 299 358 одужало, 11 263 особи померли. За даними Центру громадського здоров’я України, спостерігається такий структурний розподіл супутніх захворювань у хворих із летальністю внаслідок COVID‑19:
- 78% – серцево-судинні захворювання (ССЗ).
- 22% – ЦД.
- 13,7% – запалення легень.
- 10,5% – захворювання нирок.
- 9% – онкопатології.
- 6,4% – неврологічні захворювання.
У березні 2020 року ВООЗ назвала найчастіші хронічні захворювання в пацієнтів із COVID‑19, що зумовлюють більш високий ризик розвитку ускладнень та летальності:
- ЦД.
- Захворювання серця та судин.
- Захворювання легень.
ЦД є незалежним предиктором захворюваності та смертності в пацієнтів із COVID‑19, який може викликати різкі коливання рівня глюкози в крові пацієнтів, що негативно впливає на одужання хворих. Є підстави вважати, що декомпенсований діабет у поєднанні з пневмонією, спричиненою SARS-CoV‑2, має несприятливий прогноз. Саме тому тактика й стратегія лікування ЦД були дещо змінені, особливо за умови наявності артеріальної гіпертензії (АГ).
Основна тактика курації пацієнтів із ЦД у період пандемії COVID‑19:
- Досягнення цільового рівня глікованого гемоглобіну (HbA1c) <7%.
- Частий моніторинг рівня глюкози в крові.
- Уникнення гіпоглікемії.
- Моніторинг рівня ацетону (кожні 4-6 год) при рівні глюкози в крові >15 ммоль/л.
- Стабілізація ССЗ та хронічних захворювань нирок.
- Підтримка водного балансу.
- Правильне харчування та фізичні навантаження.
- Самоізоляція.
Микола Дмитрович наголосив, що дотепер немає даних про найбільш оптимальне лікування пацієнтів із діабетом, інфікованих SARS-CoV‑2, а також хворих на COVID‑19, в яких розвивається глікемічна декомпенсація. Жоден із препаратів не виявився домінуючим у терапевтичній стратегії лікування COVID‑19. Терапевтичний прорив було досягнуто лише при використанні дексаметазону в пацієнтів із тяжкою формою COVID‑19. Глюкокортикоїди (ГК) застосовували в пацієнтів із тяжким гострим респіраторним дистрес-синдромом як симптоматичне та протизапальне лікування. Однак використання цих засобів може погіршити резистентність до інсуліну, глікемічний контроль та спричинити виражену гіперглікемію, адже ГК проявляють свою гіперглікемічну дію, зменшуючи чутливість до інсуліну та його секрецію, а також перешкоджаючи ефектам інсуліноподібного фактору росту 1 (ІФР-1) та посилюючи вироблення глюкагону.
Лікування хлорохіном або гідроксихлорохіном може спровокувати гіпоглікемію – передусім у пацієнтів, які перебувають на терапії інсуліном або препаратами сульфонілсечовини, – через їх вплив на секрецію, деградацію та дію інсуліну. І навпаки, такі противірусні препарати, як лопінавір та ритонавір, можуть призвести до гіперглікемії та погіршити контроль глікемії. Ці засоби здатні спричинити печінкову та м’язеву токсичність, тому рекомендується бути обережним при їх застосуванні в поєднанні зі статинами та в пацієнтів із жировою хворобою печінки. Загальними є фармакокінетичні взаємодії з цукрознижувальними препаратами, що може призвести до надмірного або недостатнього впливу противірусних або протидіабетичних препаратів.
Ретельний моніторинг рівня глюкози та аналізу взаємодії лікарських засобів допоможе послабити вираженість клінічних симптомів і знизити ризики несприятливих результатів.
Хворі на діабет і COVID‑19 мають більший ризик погіршення прогнозу та смертності. Ураховуючи дуже високу поширеність діабету в усьому світі, ця когорта пацієнтів представляє значущий вразливий сегмент «ковідної популяції». Погіршення прогнозу у хворих на ЦД є ймовірним наслідком синдромного характеру захворювання: гіперглікемія, літній вік, супутні патології, зокрема ожиріння та ССЗ (у тому числі АГ). Однак насправді картина складніша, оскільки потрібно мати на увазі різні соціальні фактори. Лікареві необхідно не лише враховувати стан здоров’я хворого на ЦД, а й ретельно збалансувати цукрознижувальну терапію з конкретними методами лікування вірусної інфекції.
Знову ж таки, лікування діабету в пацієнтів із COVID‑19 – велика клінічна проблема, яка потребує комплексного підходу, оскільки це є необхідною стратегією для максимального зниження ризику медичних ускладнень та смерті.
Ретельна оцінка багатьох компонентів, здатних призвести до поганого прогнозування COVID‑19 у хворих на ЦД, може стати найкращим, якщо не єдиним, способом подолання ситуації, що склалася. Нашим системам охорони здоров’я необхідно надати можливість бути готовими до оперативного вирішення будь-яких проблем.
Нарешті, взаємозв’язок між діабетом і COVID‑19 має бути вивчений в додаткових клінічних дослідженнях, щоб зрозуміти, наскільки конкретні механізми вірусу (наприклад, його тропізм до β-клітин підшлункової залози) можуть погіршити контроль глікемії, а в деяких випадках призвести до розвитку діабетичного кетоацидозу або гіперглікемічного гіперосмолярного синдрому і, можливо, діабету.
Великий інтерес викликала доповідь віце-президента Асоціації клінічних ендокринологів України, доктора медичних наук, професора Андрія Миколайовича Кваченюка (ДУ «Інститут ендокринології та обміну речовин ім. В.П. Комісаренка НАМН України»), який докладно розповів про таку доволі рідкісну патологію, як синдром Кушинга.
Відповідно до наказу МОЗ України «Про внесення змін до Основ законодавства України про охорону здоров’я щодо забезпечення профілактики та лікування рідкісних (орфанних) захворювань» від 15.04.2014 р. № 1213-VII, усі пухлинні та гіперпластичні захворювання наднирників належать до орфанних ендокринних захворювань. Згідно зі списком, прикладеним до наказу, із 306 таких захворювань 61 нозологія – це орфанні хвороби наднирників.
Одним із орфанних захворювань є синдром Кушинга. Вісім років тому ендокринологічна світова медична спільнота відзначала 100-річний ювілей американського нейрохірурга Гарві Вільямса Кушинга, який описав синдром і тріаду Кушинга (рис. 1).
Гарві Вільямс Кушинг народився 1859 р. в Сполучених Штатах Америки. Його батьками були Бессі Вільямс і Кірке Кушинг, лікар, чия родина приїхала до Хінгема, штат Массачусетс, у XIX столітті. Гарві був молодшим із десяти дітей.
Ще дитиною Кушинг узяв участь у Клівлендській школі мануального тренування. Пізніше юнак вступив до Гарвардської вищої медичної школи. Після закінчення в 1895 р. він проходив спеціалізацію в Массачусетському головному госпіталі, а потім – у госпіталі Університету Джона Гопкінса (м. Балтимор, штат Меріленд, США) під керівництвом відомого хірурга Вільяма Стюарта Голстеда, з яким пов’язані два факти в хірургії: він рекомендував використовувати гумові рукавички при операції та описав анатомію нижніх артерій, що кровопостачають нижні паращитоподібні залози. За час своєї кар’єри Кушинг також працював хірургом у госпіталі імені Пітера Брігема в Бостоні, а також професором хірургії Гарвардської вищої медичної школи. Під час Першої світової війни (1914-1918) Кушинг деякий час провів у Франції, у військовому госпіталі, розгорнутому в Неллі, в околицях Парижа, а потім, з 1917 по 1919 р., обіймав посаду головного лікаря гарнізонного госпіталю № 5. З 1933 року і до самої смерті він працював в Єльському університеті.
Під час подорожі в Європу вивчав під керівництвом Теодора Кохера взаємозв’язок систолічного артеріального і внутрішньочерепного тиску (АТ і ВЧТ). У ході цих досліджень він спільно з Хьюго Кронекером (Hugo Kronecker, 1839-1914) виявив феномен підвищення АТ, головним чином систолічного, при збільшенні ВЧТ. Підвищення АТ грає в цьому випадку захисну роль, сприяючи посиленню кровопостачання мозку. Результати цієї роботи підштовхнули вченого до виявлення й опису рефлексу (тріади) Кушинга – синдрому, який складається з підвищеного АТ, головним чином систолічного, брадикардії (до 50-60 уд./хв) і уражень дихання при збільшенні ВЧТ. Цей синдром спостерігається при черепно-мозковій травмі, пухлинах мозку, інсульті, і зумовлений він роздратуванням життєво важливих центрів стовбура мозку.
Тільки з приводу пухлин головного мозку (підтверджених гістологічно) Кушингом було виконано понад 2 тис операцій. Він також впровадив електрокоагуляцію в нейрохірургії. Велика частина роботи була виконана спільно з доктором фізичних наук Вільямом Бов’є (William Bovie). Про важливість впровадження електрокоагуляції свідчить той факт, що до її застосування в практиці Кушинга летальність при видаленні пухлини досягала 27,7%. Після того як в клініці Кушинга стала застосовуватися «електрохірургія», летальність знизилася до 8,9%.
Найчастіше ім’я Кушинга згадується в контексті хвороби Іценка–Кушинга. 1912 р. вчений описав ендокринологічний синдром, викликаний підвищеною продукцією гіпофізом адренокортикотропного гормона (АКТГ), дав йому назву «polyglandular syndrome». Після аналізу своїх спостережень Кушинг у 1932 р. опублікував роботу «Базофільні аденоми гіпофіза і їх клінічні прояви».
Гарві Вільямс часто подорожував з академіком І.П. Павловим та спостерігав за операціями на щитоподібній залозі, які виконували Кохер і Вільярт. Пізніше він описав, що Кохер робив операцію дуже довго, тому в нього було більше випадків гіпотиреозу, а Вільярт оперував блискавично, що призводило до більш частих випадків гіпопаратиреозу. Таким чином, робить висновок Кушинг, є зв’язок між швидкістю операції і розвитком ускладнень. Бронзовий сліпок кисті «батька сучасної нейрохірургії», як часто називають видатного хірурга, сьогодні зберігається в музеї медицини Університету Джона Гопкінса як визнання досягнень Гарві Вільямса Кушинга в ендокринології та нейрохірургії.
Ендогенний гіперкортицизм
Ендогенний гіперкортицизм дуже різнопланове захворювання, яке є наслідком надмірної секреції синтезованих корою наднирників гормонів (табл. 1).
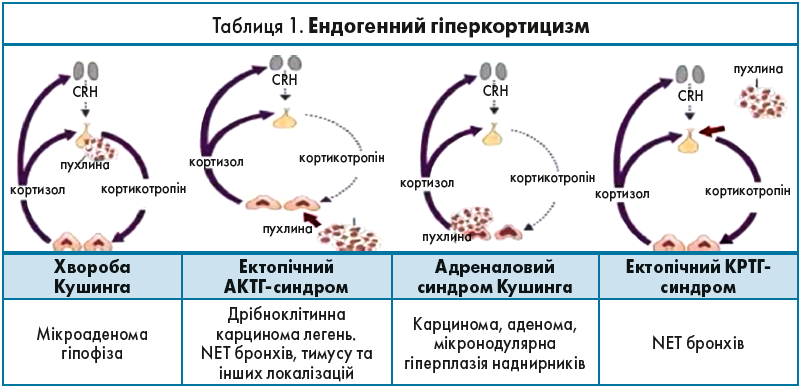
Симптомокомплекс Кушинга пов’язаний із декількома патологічними станами:
- хвороба Кушинга (причина – мікроаденома гіпофіза, гіперпродукція АКТГ);
- ектопічний АКТГ-синдром (продукція АКТГ клітинами APUD-системи, які можуть розміщуватися по всьому організму);
- адреналовий синдром Кушинга (гіперпродукція наднирниками, автономна гіперпродукція аденоми, карциноми та гіперплазії);
- ектопічний кортикотропін-релізинг-гормон (КРТГ)-синдром, також пов’язаний з клітинами APUD-системи, які розміщені в бронхах і стимулюють гіпофіз паранеопластичним процесом через виділення КРТГ та подальшої стимуляції продукції АКТГ та кортизолу в наднирниках.
Є низка захворювань зі схожою клінічною картиною, але різною етіопатогенетичною структурою. Так, розрізняють 2 основні групи ендогенного гіперкортицизму: центральна, зумовлена АКТГ, та АКТГ-незалежний синдром Кушинга (табл. 2).
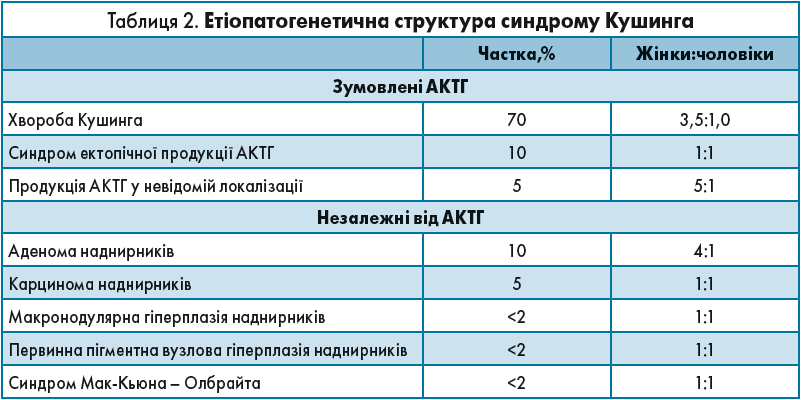
У таблиці продемонстровано, що майже 85% випадків припадає на групу, зумовлену АКТГ, і лише 15% – на незалежні від АКТГ.
Поширеність синдрому Кушинга складає (на 1 млн осіб): 55 випадків щороку – у Бельгії, 60 – у Новій Зеландії, 0,7-2,5 випадку – за даними Іспанії, Італії та Німеччини. В Україні поширеність синдрому Кушинга дорівнює 39,1 випадку на 1 млн осіб, захворюваність цією нозологією складає 2-3 випадки на 1 млн; більшість із цих випадків пов’язані з КТ-аденомою гіпофіза. Захворюваність на синдром Кушинга складає 1,2-2,4 випадку на 1 млн, а захворюваність аденомою наднирників – 0,6 випадку на 1 млн.
В Україні синдром Кушинга часто залишається недіагностованим і спостерігається:
- 2-5% випадків – у пацієнтів із діабетом на тлі ожиріння, при неадекватному контролі глікемії;
- 4,8% – у пацієнтів з ідіопатичним остеопорозом;
- 4% – у пацієнтів з АГ;
- 0,4% (1 на 250) – у жінок із гірсутизмом.
Хвороба Іценка-Кушинга – тяжке захворювання, яке супроводжується проявом великої кількісті специфічних симптомів і виникає внаслідок підвищення продукції гормонів кори наднирників, зумовленої гіперсекрецією АКТГ клітинами гіперплазованої чи пухлинної тканини гіпофіза.
За даними європейських вчених, які провели 3048 аутопсій померлих від неендокринологічних захворювань, було виявлено 334 аденоми гіпофіза, не діагностовані за життя, з них 14% становили АКТГ- продукуючі клітини.
Класична клінічна картина синдрому Кушинга
- Диспластичне ожиріння (місяцеподібне обличчя багрового кольору – «матронізм», «горб бізона»).
- Рожево-пурпурні стрії.
- Гірсутизм.
- АГ.
- Опсо-, оліго-, аменорея.
- М’язева слабкість.
- Остеопороз, патологічні переломи хребта і кінцівок.
Сучасна медична діагностика дає можливість виявити цю нозологію на ранніх стадіях, коли всі симптоми ще не проявилися і краще піддаються лікуванню. Незважаючи на це, при кожному огляді хворого необхідно звертати увагу на стан шкіри. Катаболічний ефект глюкортикостероїдів веде до витончення шкіри, аж до синдрому папірусного паперу. На рисунку 2 продемонстровано, як ендокринолог вимірює товщину шкірного покриву, щоб перевірити катаболічний вплив глюкостероїдів на товщину шкіри.
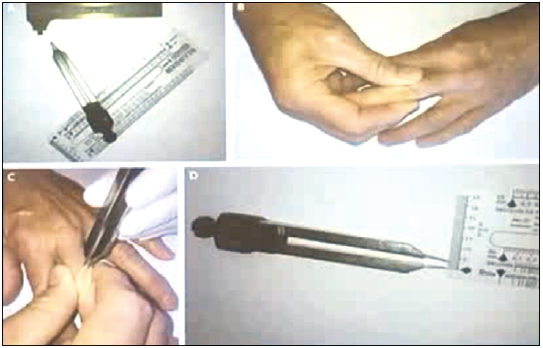
Рис. 2. Вимірювання товщини шкіри
Вченими було проаналізовано частоту клінічних проявів синдрому Кушинга (табл. 3). Найчастіше спостерігались ожиріння або характерне для цієї патології надлишкове відкладання жиру у верхній частині тулуба (95% випадків), але в пацієнта може бути синдром Кушинга і без надмірної ваги, плетори і «матронізму» (обличчя багряно-червоного кольору, іноді – з ціанотичним відтінком), без виснаження шкірних покривів (15% випадків).
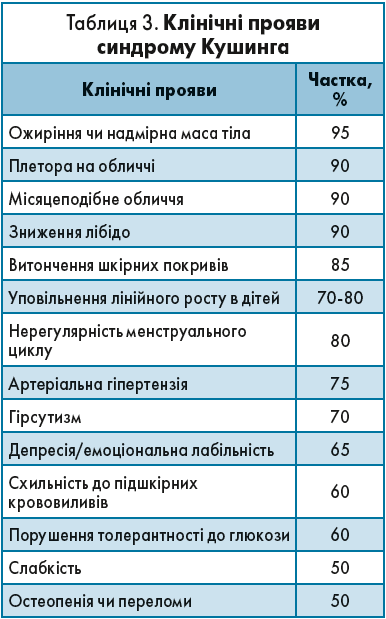
Іноді лікарі стикаються з нетиповим синдромом Кушинга, який підтверджується лабораторно. Але є когорта пацієнтів, в яких виявляються 100% усіх згаданих скарг. Це педіатрична популяція. У дітей хвороба Кушинга проявляється маніфестно і швидкоплинно.
Потрібно пам’ятати, що є поняття псевдо-Кушинг – захворювання або стани, які за зовнішніми ознаками можуть мати такий саме перебіг, як хвороба Кушинга. Зокрема:
- депресія та інші психічні порушення;
- підвищений рівень кортизолу на тлі прийому антидепресантів;
- залежність від алкоголю;
- патологічне ожиріння;
- вагітність;
- резистентність до ГК;
- неадекватно контрольований ЦД.
У цих випадках дуже важливо навіть при підвищенні кортизолу проводити дексаметазонову пробу, яка дасть можливість лікарю диференціювати вищезгадані нозології.
Біохімічні докази синдрому Кушинга наведені на рисунку 3.
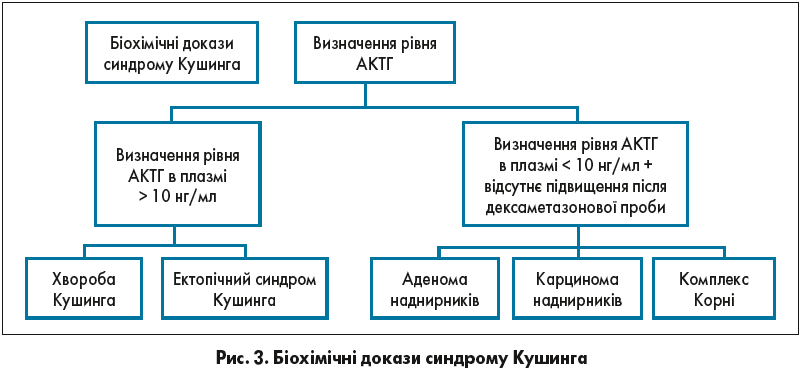
Першим кроком у разі підозри синдрому Кушинга є один із таких тестів на кортизол:
- Визначення вільного кортизолу в сечі (у 2 вимірах).
- Нічний тест із пригніченням секреції – на ніч приймають дексаметазон у дозі 1 мг (о 23:00 або 00:00 год) із подальшим вимірюванням рівня кортизолу крові о 08:00 або 09:00 годині ранку. Діагностичний критерій – концентрація кортизолу в сироватці понад 50 нмоль/л (18 нг/мл або 1,8 мкг/дл).
- Підвищені рівні кортизолу в слині пізно вввечері (>145 нг/дл (4 нмоль/л); 2 вимірювання).
- Триваліший тест із низькими дозами дексаметазону (2 мг/добу протягом 48 год) – малий тест Ліддла.
За відсутності вчасного лікування хвороби Кушинга можливий дуже несприятливий прогноз, розрахунок виживання хворих протягом 5 років складає лише 50%. При адекватному лікуванні та досягненні нормальних рівней кортизолу після двосторонньої адреналектомії виживання збільшилося до 86%.
До найчастіших причин смерті внаслідок хвороби Кушинга належать: метаболічні ускладнення, ССЗ (34% випадків), цереброваскулярні захворювання, злоякісні новоутворення, інфекційні захворювання (по 8% на кожну згадану патологію).
Основні цілі терапії хворих на синдром Кушинга:
- Нормалізація біохімічних та гормональних змін із мінімальними незворотними явищами.
- Зворотний розвиток клінічних симптомів.
- Тривалий (пожиттєвий) безрецидивний період.
За протоколом методом вибору лікування АКТГ-залежної пов’язаної з аденомою хвороби Кушинга є нейрохірургічна операція з видалення мікроаденоми (за наявності візуалізації та гістологічного підтвердження). Але варто звернути увагу на такий факт: якщо це мікроаденома, ефективність операції становить 90%, але якщо розмір аденоми дуже великий (макроаденома) – лише 65%. Зі збільшенням часу після проведення нейрохірургічної операції ризик рецидиву збільшується щороку. Наступним кроком у такій ситуації буде повторна операція, променева терапія, двостороння адренектомія або медикаментозне лікування. При виборі повторної нейрохірургічної операції ефективність втручання становить лише 50%, променевої терапії – 30-40% + розвиток пітуїтуризму. Найбільш перспективним вважають медикаментозну терапію та білатеральну адренектомію.
Медикаментозне лікування хвороби Кушинга спрямоване передусім на патогенетичну причину (звичайно – на гіпофіз), також використовують препарати, які пригнічують гіперактивність наднирників, і препарати – блокатори рецепторів ГК. Патогенетична терапія при аденомі гіпофіза – використання засобів, спрямованих на рецептори соматостатину. Аналоги соматостатину в основному експресують соматостатинові рецептори другого та п’ятого типу – sst2, sst5, агоністи дофаміну – впливають на дофамінові рецептори D2.
Дослідження М. Manavela та співавт. (2011) продемонструвало, що через 6 міс після використання каберголіну макроаденома гіпофіза значно зменшується, і коли хворий не приймає препарат, на МРТ спостерігається позитивна регресія пухлинної тканини, що свідчить про ефективність медикаментозної терапії.
Але, як показує клінічний досвід, пасіоретид, міфепристон, каберголін у монотерапії мають тимчасовий ефект. Найбільш перспективним є комбіноване лікування:
- Монотерапія пасіоретидом забезпечує 29% ефективності.
- Пасіоретид + каберголін – 53%.
- Пасіоретид + каберголін + кетоконазол – 88%, що за ефективністю можна порівняти з нейрохірургічним втручанням. Пасіоретид і каберголін блокують продукцію АКТГ аденомою, а кетоконазол інгібує стероїдогенез.
Крім того, остання комбінація препаратів дає можливість зменшити дозу кожного, а це означає менше побічних ефектів, описаних в інструкціях до цих препаратів.
Білатеральна адреналектомія – яку вважають свого роду шагом відчаю – швидко ліквідує гіперкортицизм, але потребує пожиттєвого прийому ГК. Не виключений також розвиток синдрому Нельсона (ріст аденоми гіпофіза). Цей метод використовується лише тоді, коли інші методи вже вичерпані.
Навіть після успішної терапії зберігаються:
- надмірна вага – у 50% пацієнтів;
- персистуюча гіперліпідемія та порушення толерантності до вуглеводів – у 50%;
- тривалий підвищений ризик остеопоротичних переломів;
- АГ;
- знижена якість життя.
Водночас поширеність психопатологічних змін знижується з 67% до 54%, 36%, 24% через 3; 6 та 12 міс відповідно.
Основними чинниками, які не дають подолати ускладнення, є: вік хворого; тривалість періоду, протягом якого рівні кортизолу були підвищені; вираженість гіперкортицизму; абдомінальне ожиріння; резистентність до інсуліну та атеросклероз.
Необхідно наголосити, що синдром Кушинга – комплекс специфічних симптомів, що розвиваються внаслідок дії на організм супрафізіологічних доз гормонів наднирників (насамперед ГК), а найчастішою причиною синдрому гіперкортицизму є пухлини надниркових залоз. У Брюселі (Бельгія) з 2001 р. золотим стандартом лікування доброякісних пухлин наднирників розміром менше 6 см є лапароскопічна адреналектомія. На сьогодні рамки показань ширші завдяки впровадженню цієї методики як найменш травматичної для пацієнта.
За даними Hamberger, Gajraj, Geelhoed, Siren, Crisci, поширеність інциденталоми наднирників (ІН) у світі дуже висока:
- 1% загальної популяції мають ІН;
- 4,4% – за даними КТ;
- 9% – за даними аутопсій;
- 32-73% злоякісних утворень від усіх пацієнтів з онкоанамнезом.
Більшість ІН – гормонально неактивні, але 30% із них – кортизол-секретуючі і проявляються як субклінічний синдром Кушинга. Для прийняття рішення щодо необхідності оперативного втручання лікарям потрібно визначити так званий хірургічний поріг.
Автори оцінюють його по-різному:
- ≥6 см (Ross et Aron, Lorusso).
- ≥5 см (Laronico, Osella, Outwater, Terzolo).
- ≥3 см – для хворих у віці <50 років та ≥6 см – >50 років (Staren et Prinz).
- ≥4 см (Кваченюк, Iihara, Herrera, Clark).
- ≥2 см (Brunt et Moley).
- Усі ІН <3 см підлягають ендоскопічному видаленню (Gagner, Linos).
Найбільш адекватним визначенням є розмір інциденталоми ≥ 4 см, під час операції хірург залишає, якщо є така можливість, частину нормальної тканини (приблизно 1/3). Це зберігає функцію наднирників і слугує профілактикою ризику гиперкортицизму після операції. Ризик рецидиву після залишення інтактної наднирникової тканини складає 0,5%, і якщо видалити повністю наднирникові залози, ризик смерті від адреналового кризу дорівнює 6% (1 криз із 16 закінчується летальним вислідом). Пункційну біопсію надниркових залоз проводять лише тоді, коли за допомогою клінічних та лабораторних неінвазивних методів дослідження неможливо визначити природу пухлини.
Показання до оперативного лікування при діагностуванні пухлини наднирників:
- пухлина >4 см впоперек;
- наявність ознак злоякісності при будь-яких розмірах пухлини, виявленої на УЗД, КТ (комп’ютерна томографія), МРТ (магніторезонансна томографія), ТАПБ (тонкогілкова аспіраційна біопсія);
- унілатеральна пухлина надниркових залоз після радикального лікування карциноми позанаднирникової локалізації;
- ознаки гормональної активності пухлини при будь-яких її розмірах;
- пухлина із синдромом Кушинга навіть за наявності віддалених метастазів (з подальшою хлодитан-терапією);
- неоперабельна пухлина, яка призводить до гормональної інтоксикації (з подальшою променевою чи/та хіміотерапією і хлодитан-терапією).
Адренокортикальна карцинома
З усіх пухлин ендокринної системи адренокортикальна карцинома вважається найбільш злоякісною. Ефективність її лікування складає від 10 до 60%, залежно від терапевтичної тактики. На жаль, 60% пацієнтів на момент первинної діагностики вже мають віддалені метастази, а в 40% хворих метастази розвиваються протягом перших двох років.
При діагностиці лікарю необхідно звертати увагу на розмір пухлини. Злоякісні пухлини, зокрема адренокортикальний рак (АКР), мають великі розміри на відміну від доброякісних аденом. Так що розмір є також і діагностичним критерієм з точки зору вибору операції.
Для оцінки й діагностики АКР дуже важлива топічна діагностика за допомогою КТ. Основні КТ-ознаки АКР, на які лікар має звернути увагу:
- нечіткість контурів, які характерні для АКР на відміну від чітких контурів доброякісних пухлин;
- бугриста форма;
- рівномірність контурів, що також є ознакою злоякісного процесу;
- неоднорідність структури;
- КТ-щільність. Усі злоякісні пухлини завжди мають більшу, ніж доброякісні, щільність (більш як + 30 + 51). Нульова або негативна щільність у цій нозології свідчить про доброякісний процес;
- інвазія пухлини;
- метастази пухлини в печінку, хребет, позачеревні лімфатичні вузли.
На прогноз стосовно виживання впливає, наскільки повну резекцію виконав хірург, на якій стадії було виявлено АКР і яке призначене лікування. Перший етап в лікуванні – це видалення пухлини, другий – терапія мітотаном, єдиним хіміопрепаратом, який дійсно дає можливість продовжити життя пацієнту, зменшити ризик рецидивів і полегшити його страждання. У медичному науковому журналі Fascinate проводять дослідження, де порівнюють ефективність мітотану з іншими препаратами, які призначають пацієнтові в разі непереносимості або резистентності до мітотану. За результатами випробування мітотан ефективний на будь-якій стадії пухлини, на будь-якому ступені секреції. Він насправді покращує виживаність і запобігає рецидивуванню пухлини.
Синдром Карнея
Синдром Карнея (СК) – рідкісне генетичне захворювання. Воно характеризується множинними доброякісними пухлинами (множинна неоплазія), які найчастіше вражають серце, шкіру та ендокринну систему, а також відхиленнями в забарвленні шкіри (пігменту), що призводить до плямистого вигляду на шкірі уражених ділянок. Доброякісні пухлини сполучної тканини (міксоми) часто спостерігаються в осіб із СК і, найчастіше, виявляються в серці, де вони потенційно можуть спричинити серйозні ускладнення, що загрожують життю, наприклад інсульт, обструкцію клапанів або серцеву недостатність.
При СК можуть виникати найрізноманітніші ендокринні аномалії, що вражають різні залози. До додаткових пухлин належать пухлини нервових оболонок (шванноми).
Аномалії шкірних пігментів включають крихітні плоскі (подібні до веснянок) чорні або коричневі плями (множинні лентигіни) та дрібні сині або блакитно-чорні плями (сині невуси). Специфічні симптоми та ступінь тяжкості СК можуть значуще відрізнятися в різних пацієнтів. У багатьох випадках СК зумовлений мутаціями гена PRKAR1A. Мутація може відбуватися випадково без видимих причин (тобто нова мутація) або успадковуватися як аутосомно-домінантна ознака.
СК – симптомокомплекс гіперплазії наднирників, яку іноді називають тріадою Карнея, включає в себе міксому, шкірні пігментації та гіперактивність ендокринних органів. Рак щитоподібної залози (РЩЗ) при цьому синдромі спостерігається в кожного 4-го пацієнта.
За наявності двох із наведених нижче симптомів можна встановлювати діагноз СК навіть без генетичного дослідження (рис. 4):
- серцева міксома;
- шкірна міксома;
- блакитний невус;
- депігментовані ураження;
- невус Шпіца;
- безсимптомне підвищення ГР, ІФР‑1 або пролактину;
- аденома, що продукує ГР з акромегалією;
- пролактиноми;
- міксоми повік;
- акромегілія;
- РЩЗ;
- пухлини яєчка.
Гіперкортицизм (гіперплазія наднирників) спостерігається в 45% випадків, аденома гіпофіза – у 10%, пухлини яєчка – у 56%, аденома молочної залози – у 42%, сердечні міксоми – у 72%, шкірні пігментації – у 65-70%.
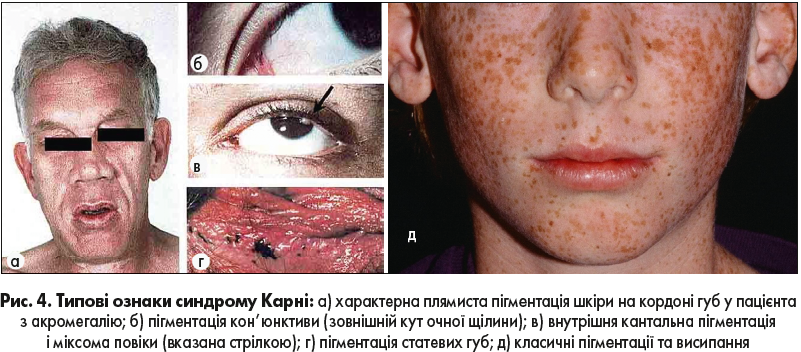
Класичні пігментації та висипання при СК не є наслідком сонячної інсоляції – це генетичні шкірні прояви.
При житті синдром Кушинга в таких пацієнтів діагностується рідко, але при розтині виявляються зміни в наднирниках, тому в разі діагностики СК з гіперкортицизмом білатеральна лапароскопічна адреналектомія є методом вибору.
Синдром MEН 1
Множинна ендокринна неоплазія 1 типу (MEН1) – рідкісний спадковий ендокринний синдром раку, що характеризується переважно пухлинами паращитоподібних залоз (95% випадків), підшлункової залози (40-70% випадків) та передньої частки гіпофіза (30-40% випадків). Фенотип MEН1 широкий, описано понад 20 різних комбінацій ендокринних та неендокринних проявів. Варто запідозрити MEН1 у пацієнтів з ендокринопатією двох із трьох характерних уражених органів або з ендокринопатією одного з цих органів плюс родич першого ступеня, уражений синдромом MEН1.
Основною причиною смерті є злоякісні пухлини підшлункової залози або карциноїд тимусу. При аутопсії в 0,25% випадків діагностується MEН1.
Синдром MEН 4
Множинна ендокринна неоплазія 4 типу (MEН4) – дуже рідкісна форма, спадковий синдром раку, що характеризується пухлинами паращитоподібної залози та передньої частки гіпофіза, можливо, пов’язаними з пухлинами надниркових залоз, нирок і репродуктивних органів.
Темою доповіді провідної наукової співробітниці ДУ «Інститут ендокринології та обміну речовин ім. В.П. Комісаренка НАМН України», кандидата медичних наук Юлії Валеріївни Булдигіної стала наднирникова недостатність – один із найтяжчих полісимптомних захворювань ендокринної системи, яке характеризується зменшенням синтезу гормонів корою наднирників (глюко- і мінералокортикоїдів) через деструктивні процеси в наднирниках різної природи.
Класифікація хронічної надниркової недостатності
- Первинний гіперкортицизм, або хвороба Аддісона, може бути вродженим чи набутим. До вроджених форм належать адренолейкодистрофія, вроджена гіперплазія кори наднирників та сімейний ізольований дефіцит ГК. Набуті форми – аутоімунний та інфекційний адреналіт, амілоїдоз, метастази в наднирники.
- Вторинний гіпокортицизм пов’язаний з ураженням гіпофіза та порушенням синтезу АКТГ. Також може бути вродженим (гіпопітуїтаризм, ізольована недостатність АКТГ) та набутим (розвивається в результаті деструктивних уражень гіпофіза пухлинами, інфекціями, крововиливами).
- Третинний гіпокортицизм пов’язаний із патологією гіпоталамуса, може бути вродженим і набутим через деструкцію тканини гіпоталамуса.
Англійського лікаря Томаса Аддісона називають батьком ендокринології. У 1855 р. він опублікував монографію, яка містила описання В12-дефіцитної анемії та хронічну надниркову недостатність (ХНН). Аддісон дуже цікавився дерматологією, і саме шкірні прояви (гіперпігментація) стали поштовхом до вивчення їх причин.
Причини хвороби Аддісона
- Аутоімунне ураження кори наднирників – спостерігається в 50-80% випадків (поєднується з іншими аутоімунними патологіями – ЦД, гіпопаратиреозом, аутоімунним тиреоїдитом, вітіліго тощо, які призводять до розвитку аутоімунного поліендокринного синдрому – одночасному ураженню декількох ендокринних залоз зі зниженням їхньої функції).
- Метастази в наднирники пухлин, розташованих в інших органах, інфекційні ураження вірусами, бактеріями, грибками, хірургічне втручання, а саме повне видалення наднирників, крововиливи в тканини наднирників через застосування антикоагулянтів.
- Туберкульоз наднирників – зазвичай більш поширений у дорослих і поєднується з туберкульозом легень.
Вторинний гіпокортицизм
Найважливіша патогенетична відмінність вторинного гіпокортицизму від первинного (первинна надниркова недостатність – ПНН) – це відсутність дефіциту альдостерону, секреція якого регулюється системою ренін-ангіотензин-Na-K.
Симптоматика вторинної надниркової недостатності (ВНН) дуже бідна: мало виражені такі симптоми, як артеріальна гіпотензія, диспептичні розлади та необхідність у солоній їжі. Важлива відмінність від ПНН – відсутність гіперпігментації шкіри й слизових оболонок. Клінічна картина ВНН включає загальну слабкість, схуднення, рідко – гіпоглікемічні епізоди. Полегшує діагностику наявність анамнестичних або клінічних даних стосовно патології гіпофіза, операційних втручань на ньому та інформація про тривалий прийом кортикостероїдів.
Діагностика ПНН
- Скарги й зовнішній вигляд хворих.
- Стійке зниження АТ.
- Погана переносимість фізичного навантаження.
- У крові пацієнтів виявляють:
- зменшення вмісту кортизолу (кров, сеча, слина);
- підвищення рівня К;
- зниження рівня Na;
- підвищення вмісту кортикотропіну (АКТГ) за законом зворотного зв’язку.
Діагностика ВНН
- Скарги та загальний вигляд хворих – без особливих змін.
- Втрата ваги.
- Зниження АТ зазвичай відсутнє.
- Погана переносимість фізичних навантажень.
- У крові пацієнтів виявляють:
- зниження вмісту кортизолу (кров, сеча, слина);
- нормальний вміст К та Na;
- зниження вмісту кортикотропіну (АКТГ).
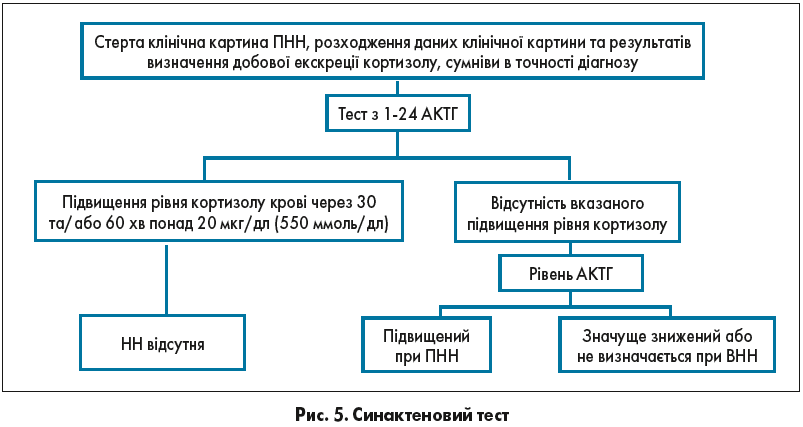
Для діагностики НН золотим стандартом вважається тест із препаратом cинактен. Синактен – це синтетичний аналог людського АКТГ, сполука, що складається з перших 24 від усього 39-амінокислотного складу природного АКТГ та має всі його фармакологічні властивості, тобто при нормальному стані кори наднирників він стимулює біосинтез кортикостероїдів.
Стимуляційний тест із дослідження рівня кортизолу плазми через 30 та 60 хв після внутрішньовенного введення 250 мкг (25 Од) синактену в розведеному в 5 мл фізіологічного розчину дає можливість дослідити тільки функціональну цілісність кори наднирників і опосередковано визначати стан всієї гіпоталамо-гіпофізарної надниркової осі, оскільки в такій дозі і за такий проміжок часу АКТГ не встигає вплинути на кору наднирника, викликаючи тільки секреторну відповідь (рис. 5).
Принципи лікування ПНН дуже часто порушуються. Треба пам’ятати, що наднирники виконують дві життєво важливі функції – це підтримка водно-сольового балансу та адаптація організму до стресорної дії зовнішнього середовища. У разі порушення першої з них, яку забезпечують мінералокортикоїди, організм гине в результаті зневоднення (втрати натрію та води) і наростаючої гіперкаліємії. Дефіцит ГК без стресорних впливів може й не позначитися.
Типовою помилкою при проведенні замісної терапії ПНН є призначення монотерапії ГК, у кращому разі кортизолом, але частіше всього – преднізолоном. У такій ситуації, незважаючи на наростаючу дозу препарату, стан хворого не нормалізується, а оскільки доза препарату збільшується, це часто призводить до розвитку екзогенного синдрому Кушинга.
Метою лікування ПНН є підібрати такий режим дозування препаратів, щоб вони відповідали фізіологічному циркадному ритму кортизолу, запобігти розвитку адреналової кризи, уникнути хронічного передозування та його віддалених небажаних ефектів (остеопороз, підвищення кардіоваскулярних ризиків, розвиток синдрому інсулінорезистентності), покращити якість життя пацієнта, забезпечивши його психосоціальну адаптацію.
У терапії ПНН еквівалентними є такі дози препаратів: гідрокортизон – 20 мг, кортизон – 25 мг, преднізолон – 5 мг (табл. 4).
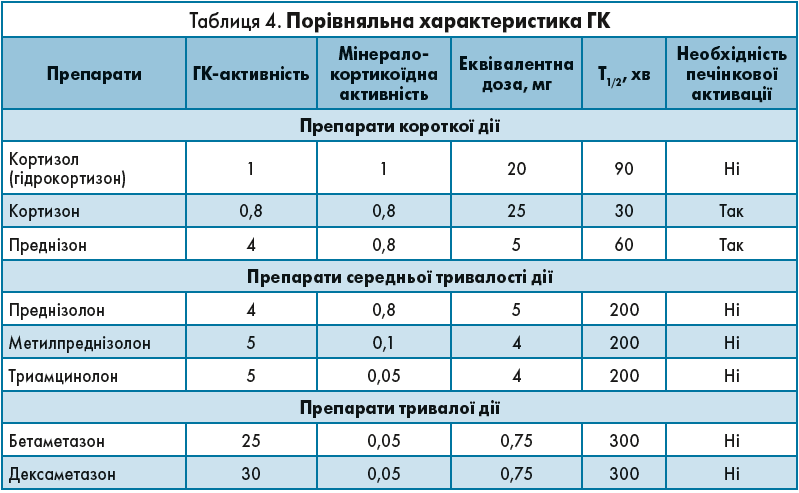
Відповідно до порівняльної характеристики ГК розрізняють такі схеми замісної терапії:
• Із використанням препаратів короткої дії:
- двохразовий режим: гідрокортизон – уранці 15-20 мг, після обіду 5-10 мг, ввечері 5 мг (чи кортизону ацетат 25; 12,5; 6,25 мг відповідно).
• Із використанням препаратів середньої дії:
- преднізолон – 5 мг зранку та 2,5 мг після обіду (значно рідше метилпреднізолон – 4 та 2 мг відповідно).
• Із використанням препаратів тривалої дії:
- дексаметазон – 0,5 мг одноразово (пізно на ніч чи вранці).
• Треба пам’ятати про циркадний ритм – максимальний викид ГК відбувається з 6-ї до 8-ї год ранку.
Паралельно лікар має проводити замісну терапію хронічної ПНН мінералокортикоїдами з використанням тільки одного препарату – 9α-фторкортизолу (флудрокортизон), який призначають 1 раз на добу в дозі 0,05-0,1 мг (щоденно, зранку); якщо спостерігається передозування, препарат приймають через день.
У клінічній практиці рекомендовано використовувати такі критерії адекватності терапії флудрокортизоном: нормальні рівні калія та натрія плазми, нормальний чи помірно підвищений рівень активності реніну плазми, нормальний (комфортний) АТ, відсутність набряків, затримки рідин (ознаки передозування препарату).
У разі призначення замісної терапії флудрокортизону вагітним необхідно передбачити деякі моменти:
- Під час вагітності відбувається поступове та значне підвищення рівня прогестерону, який є антагоністом мінералокортикоїдів.
- Унаслідок збільшення прогестерону може збільшуватися доза флудрокортизону.
- Підбір дози здійснюється з урахуванням рівня калію плазми та АТ.
- Описані випадки, коли за вказаними критеріями дозу флудрокортизону необхідно було підвищити до 0,3-0,6 мг/добу.
Оцінку адекватності замісної терапії ГК/мінералокортикоїдами проводять:
- за клінічними параметрами: збір анамнезу для оцінювання загального стану, апетиту, фізичної активності, ваги, АТ і ЧСС;
- за рівнями електролітів крові, глюкози крові натще.
Крім того, при ПНН об’єктивним критерієм компенсації мінералокортикоїдної недостатності є нормалізація рівня реніну плазми, а глюкокортикоїдної недостатності – нормалізація концентрації АКТГ у плазмі крові. Треба також мати на увазі, що визначення кортизолу в крові та сечі на тлі прийому препаратів ГК задля визначення адекватної дози не дуже інформативно й недоцільно.
Гострий гіпокортицизм
Стан гострої недостатності кори наднирників називається «аддісонічна криза», це небезпечний стан, який загрожує життю хворого. Будь-яке гостре захворювання, крововтрата, травма, операція чи інфекція поглиблює НН, що може призвести до аддісонічної кризи.
Клінічна картина аддісонічної кризи (гострого гіпокортицизму):
- блювання, діарея, що призводять до дегідратації та розвитку шоку;
- різке зниження АТ;
- втрата свідомості;
- гострий психоз чи сплутаність свідомості;
- різке зниження рівня глюкози в крові;
- гіпонатріємія, гіперкаліємія, гіперкальціємія, гіперфосфатемія.
При виникненні таких небезпечних ситуацій треба, до отримання результатів лабораторних тестів, розпочати внутрішньовенне введення 2-3 л фізіологічного розчину, можливо з 5-10% розчином глюкози. За першу добу вводять не менш ніж 4 л рідини. Введення розчинів із калієм та гіпотонічних, а також діуретиків протипоказано. Потім негайно вводять 100 мг гідрокортизону внутрішньовенно – кожні 6 год протягом першої доби (усього 500-600 мг). Як альтернативу (на час поступлення в клініку) можна ввести 4 мг дексаметазону внутрішньовенно (чи еквівалентну дозу преднізолону – 40 мг), з подальшим переходом на терапію гідрокортизоном. Паралельно проводять симптоматичну терапію. Зазвичай призначають антибіотикотерапію інфекційної/вірусної патології, яка спричинила декомпенсацію ПНН.
При позитивній динаміці доза гідрокортизону зменшується до 150-200 мг/добу на 2-гу або 3-тю добу. При стабільній гемодинаміці препарат вводиться внутрішньом’язево. Призначати мінералокортикоїди (9α-фторкортизол, кортінефф) не потрібно, доки добова доза гідрокортизону не знизиться до рівня <100 мг/добу.
Стрес, емоційні та фізичні навантаження, підвищення температури тіла, травми, хірургічні втручання можуть спровокувати підвищену потребу ГК, у цьому разі на деякий час можна збільшити дозу гормонів у 2-4 рази проти підтримувальної. При декомпенсації та/чи неможливості перорального прийому препаратів (нема апетиту, блювання, відсутність свідомості) ГК вводять парентерально – внутрішньом’язеву ін’єкцію гідрокортизону в дозі 50-75-100 мг 3-4 рази на добу з поступовим зменшенням дози і подальшим переведенням на таблетовані препарати.
При проведенні хірургічного втручання гідрокортизон вводять у дозі 50-100 мг внутрішньом’язево напередодні ввечері та в день операції зранку. Під час операції та кожні 6-8 год на добу після операції вводять 50-100 мг гідрокортизону. Внутрішньовенне введення гормонів скасовують, якщо пацієнт здатен приймати таблетовані препарати. Вагітним дозу препаратів збільшують після 3-го міс вагітності, під час пологів гормональні препарати застосовують в тому самому режимі, як при планових операціях – описано вище.
При поєднанні ХНН і гіпотиреозу спочатку компенсують НН, а потім розпочинають лікування гіпотиреозу, оскільки тиреоїдні гормони прискорюють розпад кортизолу і можуть спровокувати аддісонічну кризу.
Останньою з блоку доповідей щодо захворювань наднирників була доповідь «Феохромоцитома», з якою виступила старша наукова співробітниця ДУ «Інститут ендокринології та обміну речовин ім. В.П. Комісаренка НАМН України», кандидат медичних наук Оксана Ярославівна Самсон.
Феохромоцитома (ФХЦ) – це пухлина мозкової речовини наднирникових залоз або екстраадреналової хромаффінної тканини, яка клінічно проявляється злоякісною АГ із симпатоадреналовими кризами й різними метаболічними розладами. ФХЦ відомі також під назвою «хромафінні парагангліоми» (ПГ) і є класичними представниками пухлин APUD-системи. Злоякісні пухлини наднирників називають феохромобластомами.
Як звісно, ФХЦ секретує катехоламіни, але може продукувати й серотонін, АКТГ, вазоактивний інтестинальний пептид, соматостатин, опіоїдні пептиди, кальцитонін, пептиди з ефектами паратгормону або вазоконстрикторною дією, і тоді класична клінічна картина ФХЦ може змінюватися.
За епідеміологічними даними, поширеність ФХЦ і ПГ становить від 1-3 випадків на 10 тис населення до 1 випадку на 200 тис населення, що свідчить про надзвичайну рідкісність цієї патології. Проте з усіх виявлених таких пухлин вірогідність того, що це ФХЦ, становить 80-85% і лише в 15-20% випадків це буде ПГ. Поширеність ФХЦ/ПГ у пацієнтів з АГ складає від 0,2 до 0,6%, тобто 1 випадок на тисячу хворих на АГ. У 30-60% спостережень діагноз ФХЦ встановлюється посмертно. Пов’язують це, найперше, з ігноруванням симптомів ФХЦ, а ще, за даними літератури, у 30% випадках спостерігається латентний перебіг цього захворювання.
За результатами аутопсії ФХЦ/ПГ виявляється в 0,05-0,1% пацієнтів. У 5% пацієнтів з інциденталомою наднирників, виявленою під час анатомічного розтину, діагностовано ФХЦ. У дітей із гіпертензією поширеність ФХЦ/ПГ вище – приблизно 1,7%.
Патологічні зміни за наявності ФХЦ/ПГ у дорослих хворих: 80% випадків – це одностороння солітарна пухлина, у 10% – двосторонній процес, у 10% пухлина може бути розташована поза надниркових залоз. Відомі випадки, коли вага пухлини досягала понад 3 кг, але в більшості з них маса <100 г, а діаметр <10 см. Ці пухлини рясно васкуляризовані і отримують кров із будь-якої з трьох артерій, що постачають її в надниркову залозу. Із цих пухлин злоякісних – 10-17%, що не вдається встановити за гістологічною картиною. На злоякісність вказують тільки місцева інвазія в навколишні тканини та/або наявність віддалених метастазів у кісткову тканину та паренхіматозні органи. Злоякісність перебігу також дасть можливість встановити генетична мутація в гені, який кодує субодиницю В сукцинатдегідрогенази (SDH-B). При злоякісному перебігу ФХЦ/ПГ спостерігається більш як у 40% клінічних спостережень.
Приблизно в 10% випадків ФХЦ успадковується як аутосомно-домінантна ознака в поєднанні з деякими сімейними захворюваннями (MEН2A, MEН2B, нейрофібратоз, синдром Хіппеля-Ліндау). Проте на сьогодні найбільш поширеними є молекулярно-генетичні дослідження, і в літературі вже є дані, що у 27% пацієнтів – сімейні форми захворювання.
В успадкуванні MEN2 бере участь RET протоонкоген, який локалізується на 10-й хромосомі, мутація буде виявлена в 10-му і 11-му екзоні цього гена.
Позанадниркові ФХЦ спостерігаються вкрай рідко, їхня маса в середньому 20-40 г, а діаметр <5 см. Більшість (90%) розташована в черевній порожнині поряд із крупними судинами, у парааортальній області від діафрагми до нижніх полюсів нирок. Найчастіше (50-80%) спостерігається пухлина Цукеркандля, яка виникає з парааортального скопичення симпатичної тканини, розташованої в області відходження від аорти нижньої брижової артерії. Дуже мала частина (приблизно 1%) локалізується в області грудної клітки, у місці паравертебральних симпатичних гангліїв, 1% – у сечовому міхурі, <1% – у ділянці голови та шиї, симпатичних гангліях або позачерепних дендритах ІХ та Х черепних нервів.
ФХЦ/ПГ можуть виникнути в будь-якому віці, але найчастіше – у період від 20 до 40 років, з однаковою частотою як у чоловіків, так і в жінок. Симптоми ФХЦ/ПГ дуже різноманітні, проте наголошують на основних чотирьох, які спостерігаються у 80-90% пацієнтів – це АГ, тахікардія, головний біль, профузна пітливість. АГ погано піддається звичайному лікуванню.
Класичний перебіг ФХЦ – це пароксизмальний тип, коли на тлі нормального або підвищеного АТ розвиваються пароксизми, кризи катехоламінергічні або гемодинамічні (АТ зростає ще більше, підвищується тахікардія, підсилюється головний біль, профузна пітливість, і все це супроводжується емоційними переживаннями), пацієнти відчувають страх смерті. Криз може тривати від 1 до 5 год, може самостійно проходити, а після нього відбувається рясне сечовипускання. Також до клінічних проявів належить інциденталома надниркової залози незалежно від наявності АГ.
Початковими скринінговими методами діагностики є виявлення метанефринів плазми, визначення кортизолу сечі, або слини, або плазми крові, у разі підвищеного АТ це визначення альдостерон-ренінового співвідношення та калію плазми крові.
Треба зазначити, що симптоми ФХЦ/ПГ можуть бути спровоковані або медикаментозно (табл. 5) – після однократного прийому таких препаратів, як антагоністи допаміну, опіоїди, інгібітори моноаміноксидази (МАО), інгібітори зворотного захвату норадреналіну і серотоніну, кортикостероїди та інші, або при штучному підвищенні тиску в черевній порожнині – при пальпації, сечовипусканні, дефекації. У разі обтяженого сімейного анамнезу пацієнтові необхідно провести дослідження на наявність ФХЦ/ПГ для виключення діагнозу.
Рідше діагноз змушує запідозрити незрозуміла гіпотензія або колапс, які виникають під час хірургічного втручання або травми. Така гіпотензія розвивається через зменшення об’єму циркулюючої крові та гіповолемію.
Представлені в таблиці препарати можуть спровокувати епізод підвищення АТ у пацієнтів із наявною ФХЦ/ПГ навіть після однократного застосування. Серед цих препаратів є блокатори β-адренорецепторів: пропранолол, соталол, тимолол, надолол, лабеталол. При адреналіновому типі пухлинної секреції вони здатні спричинити парадоксальне підвищення АТ.
Показання до обстеження
- Кризовий характер АГ, особливо за наявності інших симптомів ФХЦ.
- Постійна АГ, резистентна до терапії.
- Будь-яка форма АГ у дітей.
- Однократне підвищення АТ у хворих із обтяженою спадковістю.
- Епізоди підвищення АТ після фізичного навантаження, наркозу, прийому β-адреноблокаторів або гангліоблокаторів.
- Субфебрилітет невизначеної етіології.
- Усі хворі з вперше виявленою пухлиною надниркових залоз.
Лабораторна діагностика ФХЦ
- Визначення вільних метанефринів плазми крові, фракційних метанефринів добової сечі методом рідинної хроматографії з маспектрометрією або електрохімічним аналізом.
- Висока точність методу дослідження вільних метанефринів плазми підтверджена сьогодні результатами 15 незалежних досліджень.
- Забір крові для визначення рівня метанефринів рекомендовано проводити в положенні лежачи після 30 хв горизонтального положення з використанням відповідних реверсних інтервалів.
При дослідженні фракціонованих метанефринів не завжди результати підтверджуються виявленою пухлиною. Згідно з ретроспективним аналізом, проведеним із залученням 1896 пацієнтів, у 19-21% виявлені хибно-позитивні результати внаслідок прийому препаратів (ацетомінофен, α-метилдопа, трициклічні антидепресанти, феноксибензамін, сульфасалазин, лабеталол, соталол, інгібітори МАО, симпатоміметики, кокаїн, буспірон, леводопа). Тому необхідно звертати увагу на те, які препарати пацієнт приймає і чи здатні вони впливати на показники метанефринів у плазмі та сечі.
Наступний етап діагностики ФХЦ/ПГ – це встановлення локалізації пухлини за допомогою різних методів:
- КТ – це метод вибору в топічній діагностиці ФХЦ органів грудної клітки, черевної порожнини та органів малого тазу.
- МРТ – показано пацієнтам при ПГ голови й шиї, а також у разі строгого індивідуального обмеження променевого навантаження.
- Сцинтиграфія з радіоізотопами 123І- або 131І-мета-йодбензилгуанідом (І-МЙБГ) – рекомендована за підозри на метастатичні ураження, позанадниркову локалізацію (за винятком ФХЦ/ПГ голови й шиї) або якщо ми маємо справу з рецидивуючою ФХЦ/ПГ, у разі перспективи терапевтичного використання 131І-МЙБГ.
- Позитронно-емісійна томографія (ПЕТ) з 18F-фтордезоксіглюкозою в поєднанні з КТ(ПЕТ/КТ‑18F-ФДГ) – найбільш чутливий метод при метастатичному ураженні паренхіматозних органів ФХЦ/ПГ.
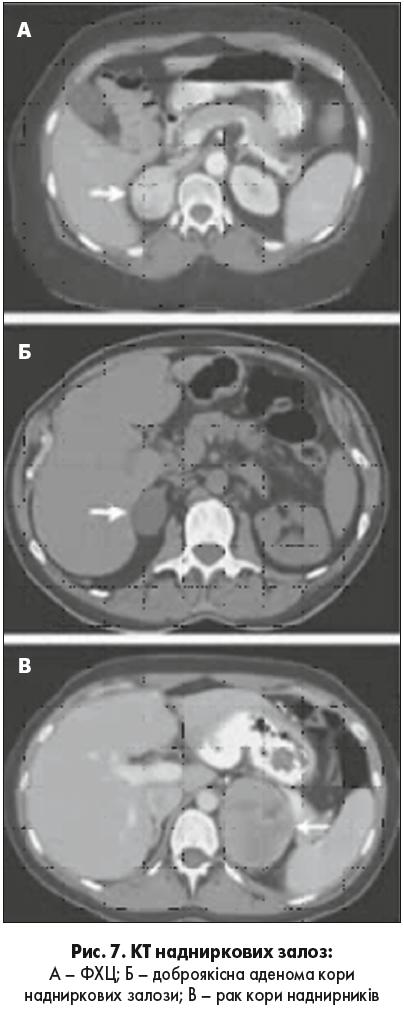
Виявити ФХЦ можна також при проведенні УЗД (рис. 6). КТ дає більш чітке та інформативне зображення. За даними КТ ФХЦ/ПГ може бути гомогенної або гетерогенної, солідної або кістозної структури, з ділянками некрозу кальцинатами (рис. 7). 87-100% пухлин ФХЦ/ПГ мають щільність понад 10 HU, відсоток «вимивання» може становити понад 60 через 15 хвилин.
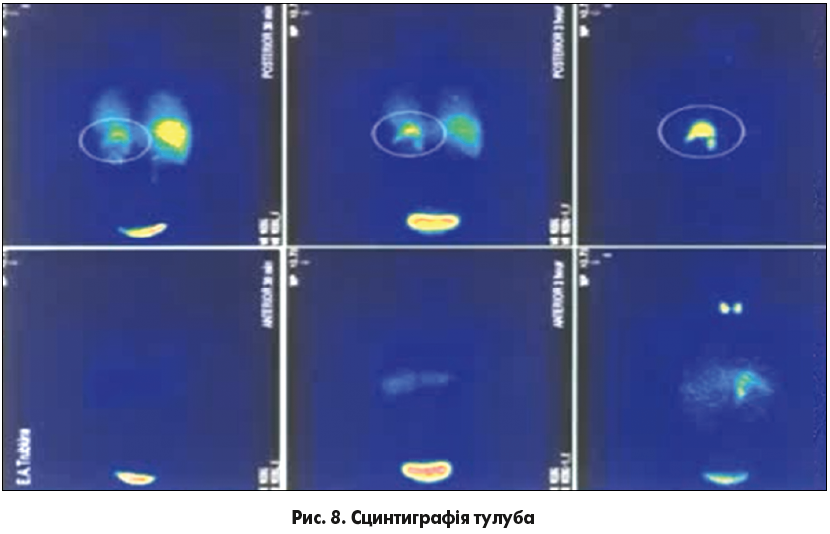
На рисунку 8 можна побачити результати сцинтиграфії тулуба в передній і задній проекції у хворого після введення 123І-МЙБГ через 30 хв, 3 год та 18 годин.
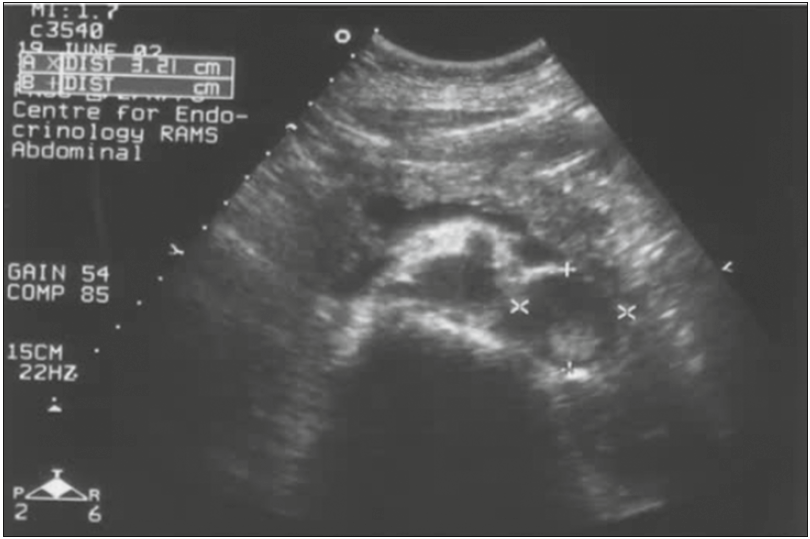
Рис. 6. Візуалізація ФХЦ на УЗД
Починаючи з 1990 року були відкриті 14 генів, чия мутація асоційована з виникненням ФХЦ/ПГ: NF1, RET, VHL, SDHD, SDHC, SDHB, EGLN1/PHD2, KIF1β, SDH5/SDHAF2, IDH1, TMEM127, SDHA, MAX та HIF2α. Це свідчить про безумовні успіхи вчених у молекулярній генетиці. У світовій медицині накопичено достатньо даних, які дають можливість зробити висновки щодо частоти виникнення, перебігу, типу секреції пухлини. У лікарів з’явилася можливість дійти певних висновків. Наприклад, пацієнтів із ПГ необхідно тестувати на мутації SDHx, а пацієнтів із метастатичними захворюваннями – на мутації SDHB. Найпоширеніша мутація в гені – SDHB (10,3%), далі SDHD (8,9%), VHL (7,3%), RET (6,3%) і NF1 (3,3%). Спадкові мутації SDHC, SDHA, MAX і TMEM127 виявлялися з частотою <2%.
Можливість таких генетичних досліджень допомагає персоналізувати підхід для ретельнішої діагностики спадкової форми ФХЦ/ПГ. Знайдені мутації генів RET і NF1 асоціюватимуться з пухлинами наднирників, які продукують переважно адреналін і метанефрин. При мутації генів VHL та SDHx зазначається переважно секреція норадреналіну та його метилованого похідного – норметанефрину. При виявленні мутації гена SDHx у більшості випадків спостерігається позанадниркова локалізація і парагангліоми голови та шиї. У пацієнтів із мутацією гена SDHB у 70% випадків визначається секреція метокситираміну – метаболіту дофаміну. Такі пацієнти мають великі позанадниркові пухлини з високим ризиком малігнізації.VHL-пухлина локалізується переважно в надниркових залозах, але в 6-15% випадків можливе поєднання з позанаднирковою локалізацією.
Візуалізація ФХЦ/ПГ буде специфічна для кожного генотипу. ПЕТ із застосуванням 18F-ФДОФА чутливіша за КТ, МРТ для виявлення парагангліом голови й шиї. ПЕТ із 18F-ФДГ – найчутливіший метод визначення локалізації будь-якої ФХЦ/ПГ, крім ПГ голови й шиї. Проведення сцинтиграфії з 111In-пентетриотидом (октреоска) є методом для виявлення ФХЦ/ПГ, асоційованих із мутацією SDHx.
Усім пацієнтам із гормонально активними ФХЦ/ПГ проводять передопераційну блокаду для запобігання періопераційним серцево-судинним ускладненням. Призначають блокатори α-адренорецепторів за 7-14 днів до проведення оперативного лікування разом із дієтою з високим умістом натрію та споживанням рідини, щоб відновити об’єм циркулюючої плазми крові та запобігти розвитку тяжкої гіпотензії після видалення пухлини.
Блокатори β-адренорецепторів варто призначати лише після досягнення α-блокади, приблизно через 3 дні, і лише якщо зберігається тахікардія. Для її усунення зазвичай достатньо низьких доз препарату.
Як можна побачити з таблиці 6, лікування починають із застосування препаратів α-адреноблокаторів – доксазозин призначають за 7-14 днів до оперативного втручання, починаючи з дози 2 мг на добу. Мета лікування – знизити АТ. Максимально дозу можна збільшити до 32 мг на добу. Якщо АТ не вдалося знизити, лікар може додати препарати другої лінії – блокатори кальцієвих каналів: ніфедипін, амлодипін. Якщо в пацієнта зберігається тахікардія, лікар має додати β-адреноблокатори.
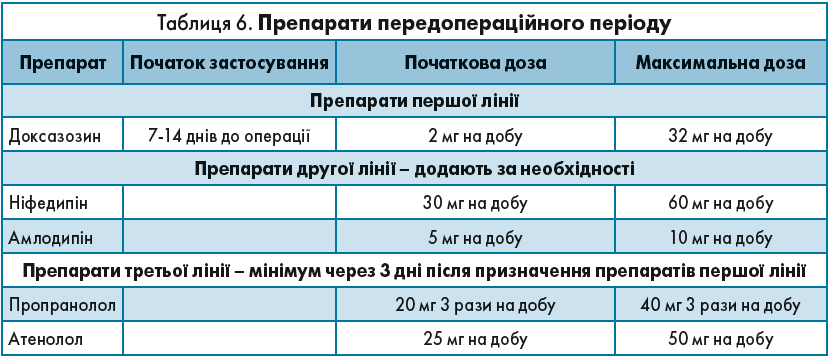
При злоякісному перебігу ФХЦ крім α-адреноблокаторів можна використовувати такий препарат, як метирозин (α-метил-паратирозин) у дозі 250 мг 4 р./добу, у разі необхідності дозу збільшують до 4 г/добу, доза регулюється за рівнем катехоламінів добової сечі та гіпотензивним ефектом; перед операцією необхідно приймати не менше 7 днів.
Але у використанні цього препарату є обмеження через деякі побічні ефекти, з яких необхідно зазначити можливість пролонгованої інтра- і післяопераційної гіпотонії, а також різні психостенічні прояви. Для впливу на пухлинний ріст при злоякісній ФХЦ/ПГ необхідно розглянути можливість хірургічного лікування, проведення радіотаргетної терапії (за наявности кісткових метастаз), а також дистанційної променевої терапії.
У передопераційний період лікарю-ендокринологу потрібно підготувати пацієнта, стабілізувати його життєво важливі показники та передати його хірургам. АТ у дорослих пацієнтів має становити <130/80 мм рт. ст. (у положенні сидячи), систолічний АТ – >90 мм рт. ст. (у положенні стоячи), ЧСС – 60-70 уд./хв (у сидячому положенні). Наявність таких показників гемодинаміки мінімізує всі ризики хірургічного втручання, яке є найефективнішим і радикальним методом лікування ФХЦ/ПГ. Необхідним обсягом операції в разі одностороннього ураження є одностороння адреналектомія, двостороннього – тотальна адреналектомія. Ендоскопічні методи проводять при розмірах пухлини до 6-8 см. Для великих (>8 см) та інвазивних пухлин кращим вважається відкритий доступ, щоб уникнути пошкодження капсули пухлини та її дисемінування. Після тотальної (двосторонньої) адреналектомії організм позбавляється ендогенних кортикостероїдів, і вже через 5-10 год після оперативного втручання розвивається недостатність надниркових залоз. Такі хворі будуть потребувати довічної замісної терапії кортикостероїдними засобами.
Після проведення хірургічного втручання потрібно провести тест на визначення метанефринів, щоб запевнитися в тому, що пухлина дійсно була солітарною і що не пропущене двостороннє ураження наднирників.
Останнім часом усе більше з’являється повідомлень про успішне лікування метастазів ФХЦ за допомогою 131І-МЙБГ. При цьому у 2/3 таких хворих спостерігається регрес розміру пухлини і збільшення тривалості життя. Виявлена експресія соматостатинових рецепторів у хромаффінних пухлинах дала унікальну можливість цільового призначення радіонуклідів (аналогів соматостатину). При злоякісних ФХЦ/ПГ експресія соматостатинових рецепторів зазначена у 80% спостережень, тому дуже багатообіцяючим є повідомлення про можливість застосування в разі поширених злоякісних форм ФХЦ радіотаргетної терапії з радіоізотопами 177 Lu-DOTA-octreotate (повна й часткова відповідь на лікування спостерігається в 35% пацієнтів, стабілізація пухлинного росту – у 75%).
Підготувала Юлія Золотухіна
Тематичний номер «Діабетологія, Тиреоїдологія, Метаболічні розлади» № 4 (52) 2020 р.
