


15 листопада, 2021
Нейропротекторні властивості тіоктової кислоти у хворих на діабет
 Діабет є одним із системних захворювань із великою кількістю ускладнень, які впливають на різні системи організму, в тому числі на серцево-судинну й нервову [1]. Зростання частки в загальній популяції літніх людей призводить до проблем, пов’язаних зі зниженням когнітивних здібностей. Відомо також, що в осіб із діабетом деменція спостерігається у 2-3 рази частіше [2].
Діабет є одним із системних захворювань із великою кількістю ускладнень, які впливають на різні системи організму, в тому числі на серцево-судинну й нервову [1]. Зростання частки в загальній популяції літніх людей призводить до проблем, пов’язаних зі зниженням когнітивних здібностей. Відомо також, що в осіб із діабетом деменція спостерігається у 2-3 рази частіше [2].
Когнітивне функціонування включає в себе кілька доменів, як-от пам’ять, мова, візуалізація, сприйняття, увага, виконавчі функції та ментальна швидкість. Пацієнти з цукровим діабетом (ЦД) 2 типу характеризуються когнітивними порушеннями порівняно зі здоровими людьми, особливо в доменах пам’яті, швидкості обробки інформації, уваги й виконавчого функціонування [3, 4]. Порушення толерантності до глюкози задовго до діагнозу ЦД 2 типу вже призводить до когнітивних порушень. Ці зміни часто відбуваються в поєднанні з іншими судинними та метаболічними розладами, включаючи гіпертонію, ожиріння й дисліпідемію, що визначається як метаболічний синдром [3].
Зв’язок між ЦД і деменцією є сильнішим для судинних когнітивних порушень, а отже, цереброваскулярні захворювання можуть бути найважливішим чинником таких порушень у разі ЦД. Хоча точні механізми, за допомогою яких ЦД впливає на мозок, залишаються незрозумілими, зміни судинної мережі мозку, порушення передачі сигналів інсуліну (Ins) у тканинах мозку, інсулінорезистентність (ІР), токсичність глюкози, окислювальний стрес, накопичення кінцевих продуктів глікування (AGE), ефекти гіпоглікемії та зміни метаболізму амілоїду можуть бути залучені до патогенезу [5, 6]. Когнітивні порушення, пов’язані з ЦД, також можуть бути опосередковані ішемією мозку, яка може мати адитивний або синергічний ефект із нейродегенеративними процесами [7].
Зміни в нервовій системі, пов’язані з когнітивними порушеннями при ЦД
1. Атрофія головного мозку визначається зменшенням тканини головного мозку, що є результатом нейродегенеративних процесів, як-от втрата нейронів і ослаблення їх взаємозв’язку. Було виявлено посилення атрофії при ЦД 2 типу, пов’язане з когнітивними порушеннями [4].
2. Хвороба церебральних малих судин (cSVD) – патологічні процеси з різною етіологією, які впливають на невеликі артерії, артеріоли, венули та капіляри головного мозку [8]. Ознаками cSVD є ураження білої речовини (WML), мікрокрововиливи, «німі» мозкові інфаркти та лакунарні аномалії [9]. Патофізіологія WML включає множинні чинники судинного (через ішемію або артеріосклероз) або запального (через транссудацію cerebrospinal fluid, CSF) походження [8]. Відзначено специфічні глибокі (підкіркові) WML і перивентрикулярні WML у пацієнтів із ЦД 2 типу, які пов’язані з порушенням когнітивної функції [9], особливо в доменах швидкості обробки, пам’яті, уваги, виконавчих функцій, а також швидкості моторики [4].
3. У пацієнтів із ЦД 2 типу виявляють «німі» мозкові інфаркти (лакунарні, кортикальні, підкіркові), які також пов’язані з порушенням когнітивних здібностей [4, 9].
4. Порушення церебральної перфузії, особливо в острівцевій корі, пов’язане з когнітивними характеристиками [10, 11]. Після введення Ins хворим на ЦД поліпшувалася пам’ять і вербальна швидкість, а також покращувалася перфузія в острівцевій корі, що свідчить про зв’язок перфузійних процесів із сигнальними механізмами Ins [4]. Ці дані вказують, що в когнітивних процесах беруть участь судинні механізми.
5. Нейрональна дисфункція належить до всіх порушень нейронної системи, включаючи зниження функціональної активності певних ділянок мозку та порушення зв’язків між ними. Дослідження з використанням амплітуди низькочастотних флуктуацій (ALFF), вимірювання спонтанної активності нейронів, регіональної однорідності, нейронної регіональної синхронізації та функціональної зв’язності показали аномальну активність мозку в пацієнтів із ЦД 2 типу [12]. Оцінка метаболічних змін кількості N‑ацетиласпартату, холіну, креатину, міо-інозитолу, γ-аміномасляної кислоти (ГАМК) і глутамату показала вищі рівні ГАМК у пацієнтів із ЦД 2 типу з високим рівнем HbA1c на тлі ослаблених когнітивних здібностей. Імовірно, в пацієнтів із ЦД 2 типу відбуваються зміни в системі ГАМК‑ергічних нейротрансмітерів, які пов’язані з когнітивною дисфункцією [4].
Механізми порушення когнітивних здібностей при ЦД
Патогенез хвороби Альцгеймера
Хвороба Альцгеймера (ХA) є нейродегенеративним розладом, пов’язаним із деградацією нейронів. Мутації в певних генах спричиняють сімейні форми ХA, але >90% пацієнтів із ХA характеризуються спорадичним типом захворювання [13].
ХA характеризується двома основними патологічними ознаками: старечими (сенільними) бляшками (SP) і нейрофібрилярними клубочками (NFT) (рис. 1) [5, 6, 14]. Ця патологія лежить в основі клінічного прояву недостачі пам’яті з подальшою втратою здатності до суджень, швидкості мови, розумових здібностей та інших когнітивних функцій. SP – позаклітинне накопичення агрегованого β-амілоїдного білка (Aβ), яке відбувається в корі головного мозку пацієнтів із ХA, що спричиняє запальні реакції шляхом активації астроглії та мікроглії [13]. Олігомери Aβ можуть індукувати синаптичну дисфункцію, порушуючи аксональне перенесення мітохондрій і везикул, що містять нейротрофічний фактор мозку (BDNF) та які відіграють вирішальну роль у синаптичній передачі [15, 16].
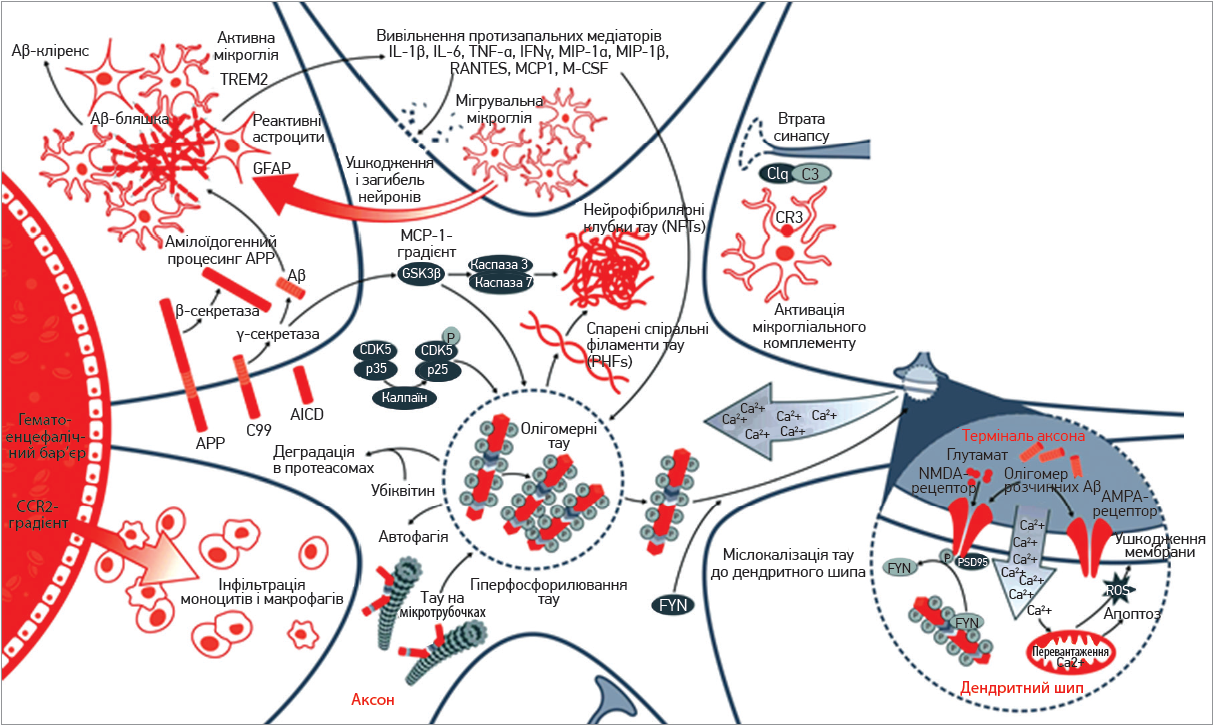
Рис. 1. Запальні процеси в нейронах і хвороба Альцгеймера (адаптовано зі схеми Abcam, Велика Британія)
На постсинаптичній мембрані олігомери Aβ взаємодіють із рецепторами глутамату та розрегульованим припливом кальцію, що погіршує довгострокове потенціювання (LTP) [13].
Патологія ЦД і Aβ
Метаболічні захворювання значно скорочують тривалість життя та пов’язані з підвищеним артеріальним тиском, серцево-судинними захворюваннями, дисліпідемією, гіперхолестеринемією й запальними станами. Дослідження показали, що в пацієнтів із ЦД 2 типу частіше розвивається когнітивна дисфункція та проявляється підвищена сприйнятливість до ХA [13, 17]. Високі рівні циркулювального Ins при ЦД 2 типу можуть чинити негативний вплив на пам’ять та інші когнітивні функції через down-регуляцію інсулінових рецепторів (IR) на гематоенцефалічному бар’єрі (ГЕБ). Як наслідок, знижується швидкість перенесення Ins у мозок, що призводить до ІР. Пригнічення передачі сигналів Ins спричиняє також порушення транспорту глюкози та її метаболізму, що сприяє змінам мітохондріальних процесів, пов’язаних із генеруванням енергії та зі збільшенням кількості реакційно-здатного кисню й азоту (ROS і RNS), які ушкоджують мітохондрії та інші клітинні компоненти [1]. Пов’язані з ІР порушення утилізації глюкози підсилюють стрес ендоплазматичного ретикулуму (ER), який дерегулює ліпідний обмін, зумовлюючи накопичення токсичних ліпідів у тканинах головного мозку. Всі ці події призводять до підвищення рівня окисного стресу, відповідального за нейродегенерацію, що спостерігається при ХA. ІР і утворення Aβ можна вважати провідними причинами підвищення окисного стресу, котрі сприяють фосфорилюванню IRS‑1/2, а також окислювальному пошкодженню білків, які беруть участь у гліколізі, циклі Кребса та синтезі АТР, що призводить до зниження метаболізму глюкози й ІР [1, 5, 6].
Патологія ЦД і тау-білок
NFT є внутрішньоклітинним накопиченням агрегованого тау-білка, пов’язаного з мікротрубочками. Гіперфосфорилювання тау індукує його агрегацію, а ступінь накопичення NFT добре корелює зі втратою нейронів і деменцією в пацієнтів із ХA [13]. У мозку мишей і щурів із ЦД 1 та 2 типів спостерігається значне збільшення вмісту фосфо-тау [18]. Однією з найважливіших протеїнкіназ, що беруть участь у фосфорилюванні тау, є GSK‑3β (кіназа глікогенсинтази‑3β) [13]. ІР може викликати аномальну активацію GSK‑3β, активність якої посилюється в моделях ЦД 1 та 2 типів, сприяючи накопиченню гіперфосфорильованого тау [19]. Ще однією протеїнкіназою, залученою до фосфорилювання тау по сайту S396 у діабетичних мишей db/db, є c-jun N‑кінцева кіназа (JNK). Активність фосфатаз також важлива для регулювання рівня фосфорилювання тау; активність деяких із них, як-от фосфатаза 2А (PP2A), порушується в тканинах мозку при ХA та ЦД [20]. Ці дані свідчать про те, що аберантний сигналінг Ins може змінювати активність GSK‑3β і PP2A, посилюючи фосфорилювання тау.
Гіперглікемія
Гіперглікемія є визнаним чинником ризику для когнітивних порушень [2]. Біологічні причини таких змін пов’язують із пошкодженням нейронів у результаті утворення AGE, окислювальним і осмотичним стресом, що руйнують ГЕБ і, як наслідок, припливом токсичних речовин, які ушкоджують нервові структури [21, 22]. Високі рівні глюкози, зміна її поглинання та метаболізму в таламусі спричиняє розвиток когнітивної дисфункції. Надлишок глюкози в мозку посилює утворення AGE, вивільнення прозапальних факторів, збільшення кількості ROS, які можуть індукувати активацію поліольних шляхів, що призводить до посилення окисного стресу й мікросудинної патології. Ці явища пов’язані зі старінням нейронів і атрофією гіпокампа [22]. Асоціація ЦД з підвищеним ризиком розвитку когнітивних порушень і деменції може бути пов’язана з периферичною гіперінсулінемією, яка впливає на кліренс Аβ й AGE. Гіперглікемія викликає електрофізіологічні зміни, а також погіршення пам’яті та здатності до навчання в центральній нервовій системі (ЦНС), що супроводжується апоптозом нейронів [23]. Судинні ускладнення в ЦНС переважно опосередковуються AGE та їхньою здатністю зв’язуватися з рецепторами – RAGE [24], що посилює окислювальний стрес і запалення, індуковані NF‑κB [23, 25]. Окислювальний стрес зумовлює загибель нейронів гіпокампа, а також зменшення кількості олігодендроцитів із подальшою втратою білої речовини. Показано зниження вмісту синаптичних білків, як-от синаптофізин і синапсин-1, фактор росту нервів (NGF), а також порушення пам’яті та здатності до навчання в результаті хронічної гіперглікемії [26].
Гіпоглікемія
У літніх людей гіпоглікемія – основне й найнебезпечніше ускладнення терапії діабету; в поєднанні з атеросклерозом призводить до органічного пошкодження головного мозку, яке часто є незворотним [5, 6, 21]. Зокрема, гіпоглікемічні події пов’язували з когнітивними порушеннями та слабоумством [22]. Найчутливішими до пошкоджувальних ефектів гіпоглікемії виявилися кора та гіпокамп. Гіпоглікемія детектується спеціалізованими нейронами, серед яких важливу роль відіграють клітини вентромедіального гіпоталамуса. Деякі дані свідчать про те, що гіпоглікемія активує шлях AMPK/Akt/GSK3, що призводить до гіперфосфорилювання тау й когнітивної дисфункції [22, 27].
Роль інсуліну й IGF. Ins регулює використання глюкози на периферії, а також метаболізм жирів і білків. Дія Ins опосередковується рецептором Ins (IR) [22, 28-30]. У мозку IR переважно локалізовані в гіпокампі (ділянка мозку, пов’язана з пам’яттю та навчанням), нюхових цибулинах, корі головного мозку, гіпоталамусі, мозочку та судинному сплетінні. Ins та IR відіграють важливу роль у навчанні та когнітивних процесах шляхом модуляції активності як збуджувальних, так і гальмівних постсинаптичних рецепторів й активації специфічних сигнальних шляхів [22]. Окрім того, Ins сприяє вивільненню Аβ у позаклітинний простір і збільшує експресію ферменту IDE [21]. Оскільки останній також руйнує Аβ, дефіцит Ins, впливаючи на кліренс Aβ, призводить до накопичення β-амілоїду [2]. Показано, що як гіперінсулінемія, так і гіперглікемія підсилюють формування нейритних бляшок [31]. Ins не тільки потрапляє через ГЕБ із периферії, але й, імовірно, утворюється локально, діючи як нейротрансмітер, регульований рівнем глюкози [32].
Ins необхідний для довгострокового потенціювання (LTP) у гіпокампі. Інфузії Ins у мозок покращували просторову пам’ять у літніх і молодих щурів [33]. Периферична гіперінсулінемія та пригнічення сигнального шляху Ins збільшують рівні Aβ і тау, що призводить до утворення SP і NFT [22].
IGF‑1 необхідний для правильного розвитку ЦНС, сприяючи нейрогенезу, синаптогенезу, розвитку та виживанню олігодендроцитів і стимулюючи мієлінізацію. Порушення його регуляції призводить до дисфункції нейронів. IGF‑1 зв’язується зі своїм рецептором із наступною активацією шляхів PI3K/Akt/mTOR і MAPK/ERK [34]. При зменшенні кількості IGF‑1 спостерігається зниження структурної складності нейронів і порушення LTP. Уведення IGF‑1 запобігає когнітивним порушенням у щурів із діабетом, окрім того, IGF‑1 регулює навчання та пам’ять і в нормальних дорослих щурів [5, 6, 22].
Отже, багато різних потенційних механізмів можуть визначати зв’язок між дисрегуляцією сигнальних шляхів Ins і розвитком когнітивної дисфункції [22]. На відміну від периферичних, рецептори Ins у ЦНС розрізняються за будовою, функцією й розміром. У людей Ins покращує когнітивні процеси незалежно від його впливу на метаболізм периферичної глюкози. Гостре підвищення вмісту Ins покращує когніцію, але хронічна гіперінсулінемія може мати негативний вплив на функцію нейронів in vitro, посилюючи сприйнятливість до токсинів і стрес-індукованих ефектів. Гліковані білки та медіатори запалення також можуть брати участь у нейродегенеративних процесах [2].
Загалом хронічна гіперінсулінемія в периферичному кровообігу поряд зі зменшенням поглинання Ins у мозку може призводити до порушення регуляції Aβ і запальних процесів.
Нейропротекторні властивості тіоктової кислоти
У 1951 р. тіоктова кислота (ТК) була визначена як найважливіший компонент енергетичного обміну й окисно-відновного стану клітин [35]. ТК (1,2-дитіолан‑3-пентанова кислота) – нейропротекторний і нейрорепараційний антиоксидант, здатний проникати крізь ГЕБ, є сильним скавенджером (поглиначем) вільних радикалів у мозку [36].
ТК синтезується мікроорганізмами, в мітохондріях тваринних і рослинних клітин. В організм людини може надходити з продуктів харчування, при вживанні темно-зелених овочів (шпинат, броколі), а також із тваринних тканин (найвища концентрація в нирках, серці та печінці) [35]. Здоровий організм людини може синтезувати достатньо ТК, що забезпечує регуляцію власних антиоксидантних і протизапальних шляхів. Однак старіння та деякі захворювання, як-от діабет, можуть зменшувати рівень ТК в організмі.
Біосинтез ТК відбувається в невеликих кількостях у мітохондріях з октанової кислоти шляхом активації синтезу жирних кислот 2 типу (FAS-II).
Потім фермент α-LA‑синтетаза поетапно та стереоселективно каталізує інсерцію атома сірки в положеннях C6 і C8 на зв’язаній із білками октановій кислоті. У клітинах людини синтез de novo жирних кислот відбувається в цитоплазмі шляхом утворення жирних кислот 1 типу та в мітохондріях за допомогою FAS-II. Синтез de novo може забезпечити обмежену кількість ТК для організму людини, саме тому ТК слід розглядати як важливий додатковий нутрієнт [35]. ТК як харчова добавка є рацемічною сумішшю, що складається з її R‑α-LA та S‑α-LA ізомерів, основні дози яких становлять 50, 100, 200, 300 або 600 мг/день. Інші дослідження показують позитивний вплив ТК у дозах 1200 та 2400 мг на особу й, що важливо, відсутність побічних ефектів. Окрім того, очищений біологічно активний ізомер R- застосовують у дозах 200 та 300 мг. Ізомер R‑α-LA нестійкий за температур >49 °C, тоді як рацемічна суміш залишається стабільною за температур від 60 до 62 °C. Фармакокінетичні дослідження, проведені на здорових суб’єктах, виявили, що ізомер R‑α-LA краще абсорбується, тоді як ізомер S‑α-LA сприяє цьому, запобігаючи полімеризації R‑α-LA. У цьому сенсі використання добавок із рацемічною сумішшю є доцільнішим [37].
У разі перорального введення ТК швидко всмоктується та виводиться через нирки. Завдяки амфіфільності ТК широко розповсюджується всіма відділами тіла, включаючи ЦНС [38]. Середній час досягнення максимальної концентрації в плазмі – 15 хв (за іншими даними, 30 хв) із періодом напіввиведення 14 (30) хв [39]. Короткий період напіввиведення ТК є результатом інтенсивної екстракції та печінкового метаболізму, що знижує біодоступність введеної дози в середньому до 30%. Різні чинники впливають на біодоступність ТК, включаючи вживання їжі, яка перешкоджає її засвоєнню. Отже, вживання ТК рекомендується за 30 хв до або через 2 год після прийому їжі [35].
Хімічна реакційна здатність ТК переважно забезпечується дитіолановим кільцем (рис. 2). У клітинах ТК відновлюється у мітохондріях шляхом NADH‑залежної реакції з ліпоаміддегідрогеназою, утворюючи дигідроліпоєву кислоту (DHLA). Як окислена, так і відновлена форми ТК мають антиоксидантні властивості та створюють потужну окисно-відновну пару. Відомо, що ТК/DHLA має окисно-відновний потенціал -320 мВ, тоді як окислювально-відновний потенціал глутатіону / окисленого глутатіону (GSH/GSSG) становить тільки -240 мВ. Ця різниця свідчить про те, що DHLA забезпечує кращий захист від окисних пошкоджень, аніж GSH. Тому пару ТК/DHLA називають універсальним антиоксидантом. Звичайні антиоксиданти є або водорозчинними, або розчинними в ліпідах мембрани агентами. Натомість ТК має як гідрофільні, так і гідрофобні властивості. Цей амфіфільний характер ТК є унікальним серед антиоксидантів. Насправді окислювально-відновна пара ТК/DHLA здатна регенерувати кілька антиоксидантів, і, на відміну від аскорбінової кислоти, DHLA не руйнується під час гасіння вільних радикалів і може бути відновлена з ТК [40]. Існують також докази, що ТК підвищує або підтримує рівень клітинного GSH, діючи як індуктор транскрипції генів, що регулюють його синтез. Лікування ТК підвищує рівень ядерного Nrf2 у печінці й індукує транскрипцію генів, опосередковану Nrf2. Nrf2 є ключовим фактором транскрипції, який опосередковує експресію генів антиоксидантів і детоксикації, регульованих елементом антиоксидантної відповіді (ARE). Показано, що ТК також пригнічує активацію NF‑κB – ключового прозапального фактора, пов’язаного із секрецією запальних цитокінів. Застосування ТК у клініці значною мірою пов’язане з дією ТК на сигнальний шлях інсуліну. Вона стимулює поглинання глюкози шляхом регулювання транслокації й активності транспортерів глюкози – ефект, який може бути опосередкований р38-мітоген-активованою протеїнкіназою (p38MAPK) [40].
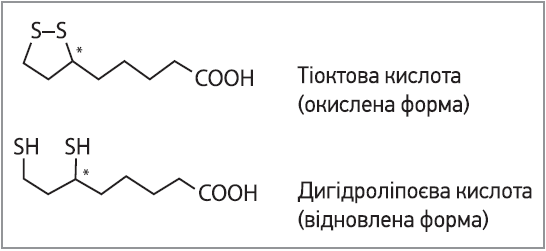
Рис. 2. Структура окисленої та відновленої форм тіоктової кислоти
Судинна деменція (СД) характеризується набутими когнітивними порушеннями, які виникають внаслідок цереброваскулярних захворювань. СД зазвичай пов’язана з порушеннями мозкового кровообігу, як-от інсульт, а чинники ризику включають гіпертонію, ЦД, метаболічні розлади та метаболічний синдром. Цереброваскулярні захворювання зменшують кровопостачання мозку й підвищують окислювальний стрес у мозковій тканині, послідовно спричиняючи гіпоксію, аноксію та запальні реакції. Крім того, цереброваскулярні захворювання призводять до втрати вегетативного контролю судин, функціональних порушень капілярів і пошкодження білої речовини та гіпокампа, що з часом призводять до симптомів СД [41].
Хоча СД характеризується найвищим рівнем захворюваності після ХА серед синдромів деменції, нині не існує надійних терапевтичних фармакологічних методів лікування. Клінічне лікування СД зосереджується лише на застосуванні різних препаратів для запобігання подальшому когнітивному погіршенню. Серед цих препаратів ТК використовують як потужний антиоксидант. ТК елімінує активні форми кисню, сприяє виробленню глутатіону та відновлює антиоксиданти, як-от вітаміни Е та С, таким чином виконуючи важливу роль в антиоксидантній мережі організму [42]. Оскільки ТК легко всмоктується й перетинає ГЕБ, доведено, що вона захищає від нейродегенерації та невропатичного болю [43]. У гризунів із двобічним стенозом загальної сонної артерії в гіпокампі спостерігали когнітивну дисфункцію й апоптоз, опосередковані PTEN/Akt/mTOR. Діючи на шлях mTOR α-LA покращувала когнітивну функцію та сприяла виживанню клітин гіпокампа [41]. Отже, ТК може бути корисною для лікування СД.
ТК захищає мозок від реперфузійних травм на ранніх стадіях ішемії мозку. Показано, що лікування щурів у стані оклюзії церебральної артерії ТК захищає мозок від ішемічного інсульту шляхом модуляції декількох запальних шляхів. Окрім того, лікування пацієнтів протягом 3 тиж ТК забезпечувало значне зниження рівнів прозапальних чинників – інтерлейкіну-6 (IL‑6) і фактора некрозу пухлини (TNF) порівняно з вихідними показниками [36].
Нейропротекторні ефекти ТК при ішемічній травмі було продемонстровано в декількох різних експериментальних моделях. Одержані дані свідчать про те, що антиоксидантні властивості ТК, зокрема здатність відновлювати вміст GSH, корелюють з її здатністю сприяти гліально-нейрональній взаємодії та нейропроліферації. Ці результати вказують на те, що негайне лікування ТК після ішемічної травми може мати значні нейрорепаративні ефекти, опосередковані (щонайменше частково) активацією рецепторів інсуліну. Отже, ТК може бути корисною для лікування гострого ішемічного інсульту [44].
Тобто ТК діє як потужний нейропротектор, сприяючи нейропроліферації після ішемічного ураження мозку. Незважаючи на доступність найсучасніших нейрохірургічних методів, інфаркт ділянки середньої мозкової артерії все ще призводить до високого рівня смертності. Отже, нейрорепаративні шляхи та шляхи виживання є потенційними терапевтичними мішенями при гострій ішемічній травмі в клінічних умовах. Термінове лікування ТК, яке проводиться після інсульту протягом короткого вікна лікування, має значний нейрорепаративний ефект і сприяє довгостроковому функціональному відновленню завдяки посиленим протизапальним й антиоксидантним діям, опосередкованим (щонайменше частково) активацією рецепторів інсуліну. Ці результати надають додаткові докази позитивних терапевтичних ефектів ТК при лікуванні ішемічного інсульту й дають змогу розробляти нові клінічні підходи щодо мінімізації пошкодження внаслідок ішемії/реперфузії [44].
Для дослідження нейропротекторних ефектів ТК використовували модель оклюзії/реперфузії середньої мозкової артерії. Показано, що ТК послаблює пошкодження внаслідок церебральної ішемії та реперфузії через IR/Akt-залежне інгібування NADPH‑оксидази. Встановлено, що ТК може активувати рецептори інсуліну та сигнальні шляхи PI3K/Akt, інгібувати експресію й активність NADPH‑оксидази, а згодом пригнічувати утворення супероксиду та знижувати ключові показники окисного стресу, включаючи малоновий діальдегід, карбонілювання білка та 8-OHdG (8-hydroxy‑2’-deoxyguanosine). ТК також знижує кількість TUNEL‑позитивних клітин і посилену активність каспази‑3, індуковану за умов оклюзії/реперфузії середньої мозкової артерії [45].
Останнім часом досліджується вплив ТК на захворювання ЦНС, як-от ХА, хвороба Паркінсона (ХП), хвороба Гантінгтона (ХГ), розсіяний склероз (РС) і пошкодження спинного мозку.
Дисфункція мітохондрій відіграє важливу роль у патогенезі інвалідизувальних нейродегенеративних захворювань, як-от ХА, ХП, ХГ, РС, аміотрофічний латеральний склероз й атрофія м’язів хребта, через порушення біоенергетичних властивостей клітин [46]. На сьогодні звичайні терапевтичні препарати мають низку обмежень через різноманітність клітинних сигнальних шляхів, задіяних у разі цих хвороб, і токсичний потенціал цих засобів. ТК – це жирна кислота, котру використовують як добавку при низці захворювань, як-от периферичні невропатії та нейродегенеративні розлади. ТК діє як ферментативний кофактор, здатний регулювати обмін речовин, вироблення енергії та біогенез мітохондрій, що дало змогу назвати її мітохондріальним нутрицевтиком. Антиоксидантна здатність ТК пов’язана з двома тіоловими групами, які запобігають надмірному утворенню вільних радикалів і поліпшують функцію мітохондрій. Ба більше, ТК впливає на механізми епігенетичної регуляції в генах, пов’язаних з експресією різних запальних медіаторів, як-от PGE2, COX‑2, iNOS, TNF, IL‑1β та IL‑6. Що стосується фармакокінетичного профілю, то ТК характеризується швидким поглинанням, низьким ризиком у разі тривалого застосування та невеликою біодоступністю, а метаболізується головним чином у печінці [37].
Пероральний довгостроковий прийом ТК до 60 мг/кг/день у щурів не виявив негативних наслідків щодо маси тіла, гістопатологічних висновків й аналізів крові. Клінічні випробування з оцінкою несприятливих наслідків для здоров’я людей проводили в дозах до 2400 мг/день, які показали відсутність побічних негативних ефектів проти плацебо [47].
Було показано, що тривале лікування ТК зменшує залежний від гіпокампа дефіцит пам’яті, значно покращуючи навчання та пам’ять у водному лабіринті Морріса, порівняно з мишами Tg2576 (модель ХА), які не отримували ТК [48]. В іншому дослідженні оцінювали ефекти ТК на мишей SAMP8 (senescence-accelerated mouse prone 8) із порушенням навчання та пам’яті, які показали, що ТК може покращити пам’ять у різних парадигмах. Досліди щодо розпізнавання об’єктів продемонстрували, що миші, які отримували ТК, мали вищий індекс пам’яті, ніж контрольні миші [49].
У трансгенних мишей зі зміною тауопатії знижували рівень білка тау, модулюючи активність кальпаїну-1 і, можливо, інших кіназ. Окрім того, спостерігали припинення втрати нейронів і фероптозу за дії ТК.
Цікаво, що за нейропсихологічною оцінкою, загальний рівень деменції при лікуванні ТК покращувався в пацієнтів, які мали ХА та ЦД 2 типу, порівняно з групою лише з ХА на всіх етапах дослідження [37].
ХП є другим за частотою нейродегенеративним розладом у людей, що старіють, і характеризується руховими симптомами, пов’язаними зі втратою дофамінергічних нейронів у чорній субстанції, що призводить до зменшення дофамінергічних терміналів смугастого тіла. Дослідження показали, що, крім зменшення когнітивного дефіциту, ТК також може ослабити рухові порушення, пов’язані з ХП [50]. ТК забезпечувала нейропротекцію від втрати нейронів чорної субстанції, що може бути результатом ослаблення окисного стресу, а також підвищувала рівень дофаміну та норадреналіну в гіпокампі щурів навіть у дозі 20 мг/кг [35].
ХГ – хронічне нейродегенеративне захворювання та спадковий автосомно-домінантний розлад ЦНС, спричинений єдиною генетичною мутацією [51], що характеризується загибеллю нейронів у хвостатому ядрі, зовнішній частині сочевичного ядра мозку, корі головного мозку, меншою мірою – в гіпокампі та ядрі субталамуса [52]. Цей розлад характеризується руховими симптомами та когнітивними й поведінковими особливостями [51]. Дослідження ефектів ТК щодо ХГ у щурів показало, що лікування α-LA покращувало просторову пам’ять, оцінену за допомогою водного лабіринту Морріса [52]. Автори продемонстрували, що добавки α-LA впливають на просторову пам’ять, послаблюючи окислювальну травму, спричинену іонами заліза та міді, що спостерігається при вікових розладах.
РС – це захворювання, котре призводить до втрати мієліну в ЦНС. Активні форми кисню (АФК) є токсичними метаболітами. Накопичені дані вказують на те, що опосередкований АФК апоптоз олігодендроцитів (OLG) відіграє важливу роль у патогенезі РС за умов окисного стресу [53]. Дослідження на експериментальній моделі РС впливу ендогенного антиоксиданта ТК як скавенджера АФК щодо загибелі OLG та дегенерації мієліну під час демієлінізації, спричиненої купризоном, показали, що лікування ТК значно збільшувало популяцію зрілих OLG (клітин MOG+), а також знижувало окислювальний стрес (за рівнем АФК, COX‑2 і PGE2) та кількість медіаторів апоптозу (за рівнем каспази‑3 та співвідношенням Bax/Bcl2) у мозолистому тілі. ТК значно стимулює популяцію клітин глії NG2+, або полідендроцитів (NG2-хондроїтинсульфат-протеоглікан-позитивна глія) з 4-го тижня. Отже, ТК може запобігати апоптозу, затримує демієлінізацію та стимулює механізми виживання й регенерації OLG у мозолистому тілі [54]. Показано також, що вживання 1200 мг ТК на добу сприятливо впливає у хворих на РC на кілька запальних цитокінів, включаючи INF‑γ, IL‑4, TGF‑β та молекули адгезії ICAM‑1, VCAM‑1 [35, 55].
Уведення ТК тваринним моделям із РС значною мірою зменшує запалення, демієлінізацію та втрату аксонів, а також зменшує кількість CD3+ Т‑клітин та CD11b+ моноцитів/макрофагів у спинному мозку. Було встановлено, що лікування ТК значно зменшувало кількість CD4+ та галектин‑3+ імунних клітин у мозку [56]. LA може виявитися корисним у лікуванні РС, пригнічуючи активність MMP‑9 і перешкоджаючи міграції Т‑клітин у ЦНС [35].
Отже, ТК проявляє захисний ефект проти токсичної демієлінізації за рахунок гальмування окислювально-відновного сигналінгу та зменшення вразливості полідендроцитів до ексайтотоксичної дії.
В іншій роботі досліджувалися нейропротекторні ефекти ТК за умови нейротоксичності, викликаної введенням AlCl3 мишам [57]. ТК посилювала пам’ять про пережитий страх і перевагу соціальної новизни порівняно з групою, яка отримувала тільки AlCl3. Було показано, що ТК нейтралізує дію токсичних металів на нервову тканину як безпосередньо, так і побічно завдяки своїй здатності підвищувати рівень внутрішньоклітинного глутатіону. На додаток до прямих антиоксидантних властивостей виявилося, що ТК і DHLA мають здатність хелатувати активні в окисно-відновлювальних реакціях метали, як-от мідь, вільне залізо, цинк [35].
ТК є унікальною серед природних антиоксидантів сполукою, адже вона відповідає багатьом вимогам, що робить її потенційно високоефективним терапевтичним засобом у разі патологічних станів, пов’язаних з окисними пошкодженнями. Є дані, що ТК демонструє терапевтичний ефект навіть щодо зниження рівня глюкози в умовах діабету, стимулюючи експресію та мобілізацію на клітинну мембрану транспортерів глюкози. Так само добавки ТК мають численні сприятливі ефекти на відновлення функції мітохондрій і на окислювальний стрес, пов’язаний із деякими захворюваннями та старінням [58].
Показано, що сигнальний шлях PI3K/Akt, який є ключовим щодо регуляції росту клітин, проліферації, диференціації, виживання та метаболізму, модулюється ТК. Зокрема, дисбаланс PI3K/Akt, пов’язаний із віком і ХА, відновлювався в щурів протягом 3 тиж після додавання ТК у питну воду [59]. Лікування ТК значно посилює фосфорилювання Akt і призводить до відновлення співвідношення pJNK/pAkt у корі мозку щурів при старінні. Запобігання індукованого севофлураном апоптозу за допомогою ТК відбувалося шляхом відновлення рівнів фосфорилювання Akt і GSK3-β у гіпокампі [60]. Активуючи сигнальний шлях PI3K/Akt, ТК, яку вводили протягом 3 днів, поcлаблювала ішемію головного мозку та зменшувала пошкодження, спричинені реперфузією, в дорослих щурів [45, 48].
Тобто проведені дослідження переконливо свідчать про безпосередню роль антиоксидантних механізмів дії ТК у реалізації її клінічних ефектів. Слід зазначити, що в ангіоневрології на сьогодні для дуже малої кількості антиоксидантів є переконлива доказова база, що свідчить про кореляцію клінічних і біохімічних параметрів їхньої ефективності, тобто верифіковане підтвердження клінічної значущості їхніх біохімічних ефектів. ТК у цьому відношенні має пріоритет перед більшістю сучасних нейропротекторів-антиоксидантів, що дає можливість обґрунтовано говорити про її реальні клінічні можливості. Загалом сьогодні ТК може розглядатись як один із найперспективніших нейропротекторів у терапії різних форм гострих і хронічних порушень мозкового кровообігу. З огляду на значення вільнорадикальних процесів у механізмах старіння мозку в розвитку нейродегенеративної патології (ХА та ХП) [61] можна припускати також доцільність застосування цього засобу в лікуванні дозозалежних форм неврологічної патології, а також пов’язаних із віком когнітивних розладів (синдром м’якого когнітивного зниження).
Сьогодні на українському ринку продуктів ТК представлена дієтична добавка Тіокт Q10 в інноваційній формі контрольованого вивільнення активної речовини. Це дає можливість постійної підтримки необхідного рівня ТК в організмі людини.
Тіокт Q10 вироблений за унікальною запатентованою технологією M.A.T.R.I.S.®, яка забезпечує покриття частинок ТК інноваційними полімерними мембранами. Саме така форма забезпечує контрольоване вивільнення активної речовини протягом 8 год від моменту прийому. Унікальна технологія дає змогу значно розширити межі фармакокінетики ТК для повнішого використання її терапевтичних властивостей. У Тіокт Q10 це відбувається шляхом реалізації механізму FAST/RETARD, тобто частина активної речовини вивільняється швидко (FAST), а друга частина – повільно й постійно (RETARD), що гарантує максимальну ефективність [62].
Неможливо не звернути увагу на ще одну важливу особливість Тіокт Q10 – високе дозування активної речовини: 800 мг ТК. Це дає змогу заповнити нейрональний дефіцит значно швидше порівняно з іншими препаратами ТК [63].
На окрему увагу заслуговує лікарська форма саше, що значно підвищує безпеку та прихильність до лікування. Окрім ТК, дієтична добавка Тіокт Q10 містить іще два компоненти: ацетил-L‑карнітин (1000 мг) і коензим Q10 (25 мг).
Ацетил-L‑карнітин поліпшує енергетичний статус клітин, сприяючи β-окисленню жирних кислот на мітохондріальному рівні, а також ефективно протидіє окислювальному стресу та пригнічує експресію прозапальних медіаторів [64].
Коензим Q10, своєю чергою, забезпечує захист від перекисного окислення ліпідів як на клітинному, так і на мітохондріальному рівні, запобігаючи нейрональній загибелі, а також знижує синтез прозапальних молекул [65].
Отже, Тіокт Q10 – це ретельно підібрана комбінація складових діючої речовини, яку слід рекомендувати для підтримки нормального функціонування органів, тканин і клітин, а також як джерело енергії для функціонування та забезпечення життєдіяльності організму.
Висновки
ТК – потужний природний антиоксидант, який впливає на низку клітинних процесів, включаючи пряме видалення радикалів, рециклінг, хелатування металів, регенерацію ендогенних антиоксидантів, як-от вітаміни С та Е, модуляцію активності факторів транскрипції, пов’язаних з експресією клітинних антиоксидантів. ТК діє також як кофактор біоенергетичних мітохондріальних ферментів. Показано, що вона покращує функцію ендотелію та кровоток, відновлює пошкоджені клітини в ЦНС, а також прискорює синтез GSH, який відіграє вирішальну роль у регулюванні експресії декількох антиоксидантних і протизапальних генів, має протидіабетичні властивості.
Сучасні дані надійно свідчать про те, що ТК характеризується потужними нейропротективними ефектами та здатна покращувати когнітивні параметри, усуваючи порушення, пов’язані з різноманітними нейродегенеративними розладами (ХА, ХП, ХГ, РС, пошкодження спинного мозку та ін.), впливом нейротоксинів, металів, а також із нормальним старінням. ТК покращує пам’ять у різних парадигмах навчання та пам’яті, включаючи аверсивну, просторову й розпізнавальну пам’ять.
В основі біохімічних механізмів дії ТК лежать її позитивні ефекти щодо інсулінового рецептора, сигнального шляху PI3K/Akt і рівня Nrf2, а також пригнічення ключового прозапального фактора NF‑κB, експресії та секреції запальних цитокінів і медіаторів, як-от PGE2, COX‑2, iNOS, TNF, IL‑1β та IL‑6.
Список літератури знаходиться в редакції.
Соколова Л.К., Пушкарьов В.М., Тронько М.Д. Нейропротекторні властивості α-ліпоєвої кислоти у хворих на діабет. Проблеми ендокринної патології, 2021; 78 (4): 146-158.
UA-TIOK-PUB-112021-037
Медична газета «Здоров’я України 21 сторіччя» № 19 (512), 2021 р.
