


16 січня, 2022
Цукровий діабет і статини
 Цукровий діабет (ЦД) вважають еквівалентом хронічного коронарного синдрому (ХКС) і чинником ризику гострих коронарних синдромів (ГКС). Інтенсивний контроль глікемії при ЦД 1 та 2 типів сприяє значному зниженню рівня мікросудинних ускладнень, частоти ГКС, смертності внаслідок ускладнень серцево-судинних захворювань (ССЗ). Однак результати досліджень ACCORD і ADVANCE свідчать, що агресивний інтенсивний контроль глікемії, досягнення та підтримання довготривалої жорсткої нормоглікемії чинять потенційно шкідливі ефекти на перебіг ССЗ у хворих на ЦД.
Цукровий діабет (ЦД) вважають еквівалентом хронічного коронарного синдрому (ХКС) і чинником ризику гострих коронарних синдромів (ГКС). Інтенсивний контроль глікемії при ЦД 1 та 2 типів сприяє значному зниженню рівня мікросудинних ускладнень, частоти ГКС, смертності внаслідок ускладнень серцево-судинних захворювань (ССЗ). Однак результати досліджень ACCORD і ADVANCE свідчать, що агресивний інтенсивний контроль глікемії, досягнення та підтримання довготривалої жорсткої нормоглікемії чинять потенційно шкідливі ефекти на перебіг ССЗ у хворих на ЦД.
Основні порушення ліпідного обміну при цукровому діабеті
Відомо, що провідна роль у патогенезі ССЗ при ЦД належить механізмам, пов’язаним із хронічною гіперглікемією і діабетичною (атерогенною) дисліпопротеїнемією (ДЛП) (гіпертригліцеридемією, гіперТГ), низьким рівнем холестерину ліпопротеїнів високої щільності (ХС ЛПВЩ), збільшенням концентрації малих та щільних частинок ХС ліпопротеїнів низької щільності (ХС ЛПНЩ). Повідомляється, що ефективне лікування ДЛП, артеріальної гіпертензії супроводжується зниженням частоти макросудинних ускладнень. Отже, зменшення ризику розвитку ССЗ у хворих на ЦД потребує багатофакторного підходу, зокрема, контролю провідних атерогенних чинників.
Збільшення рівня ХС ЛПНЩ відповідає за відносний ризик ССЗ у >50% населення. Однак ХС ЛПНЩ – не єдиний представник ліпідів, який визначає цей ризик; зокрема, в пацієнтів із ЦД 2 типу, метаболічним синдромом (МС) та/або верифікованими ССЗ переважає атерогенна ДЛП, що характеризується підвищеною концентрацією в крові ТГ, ліпопротеїнів, збагачених ТГ, часто з підвищеним вмістом аполіпопротеїну B (apoВ) і атерогенного ХС (загальний ХС (ЗХС) мінус ХС ЛПВЩ) і низьким вмістом ХС ЛПВЩ, а також аполіпопротеїну А-І (аpоА-І). До інших асоційованих чинників ризику належать збільшення концентрації постпрандіальних ТГ, кількості часток ЛПНЩ, аполіпопротеїну С-ІІІ (аpоС-ІІІ). Рівень ХС ЛПНЩ у пацієнтів із ЦД та/або МС часто знаходиться в межах фізіологічної норми чи незначно підвищений, а вміст аpоВ може бути збільшеним. Відомо, що концентрація аpoB – показник суми вмісту атерогенних часток – дуже низької щільності, ліпопротеїнів проміжної щільності (ЛППЩ), ремнантів ЛППЩ, ЛПНЩ і ліпопротеїну (а). Не дивно, що зміни концентрації аpоВ є кращим індикатором ризику ХКС, ніж ХС ЛПНЩ, а також тісніше пов’язані з компонентами МС. Маленькі щільні частки ЛПНЩ зі зниженим вмістом ХС зазвичай асоціюються із МС; водночас вони не є незалежним предиктором ХКС (за винятком ТГ, ХС ЛПВЩ або apoB). АpоА-І і аpоА-ІІ, провідні аполіпопротеїни ЛПВЩ чинять потенційні атеропротекторні ефекти та сприяють зменшенню ризику ССЗ. Водночас аpоC-ІІІ, що входить до складу деяких ліпопротеїнів дуже низької щільності (ЛПДНЩ) і ЛПНЩ, збагачених ТГ, також є сильними незалежними предикторами ССЗ. Зокрема, аpoC-III може належати патофізіологічне значення при ЦД: причетність до дисфункції β-клітин і розвитку мікросудинних ускладнень, безпосередньої активації прозапальних, а також атерогенних механізмів в ендотеліоцитах/моноцитах. Повідомляється, що в пацієнтів з атерогенною ДЛП, гіперТГ, МС, інсуліновою резистентністю (ІР) і ЦД 2 типу спостерігається збільшення концентрації apoC-III в крові. Атерогенні ДЛП також пов’язані з прозапальним статусом, який, своєю чергою, вносить вагому частку щодо ризику розвитку ССЗ. Зокрема, частки ЛПДНЩ, збагачені ТГ, активують ядерний фактор каппа-В, якому належить ключова роль в активації спектра прозапальних генів, що сприяє ендотеліальній дисфункції та оксидативному стресу (ОС). ЛПДНЩ і ЛПНЩ при ЦД характеризуються підвищеною сприйнятливістю до ліполізу, що супроводжується збільшенням концентрації неетерифікованих жирних кислот і лізофосфатидилхоліну в ліпопротеїнах, розвитком прозапального статусу. Збільшення рівня системних маркерів хронічного запалення, зокрема високочутливого C-реактивного протеїну та запальних цитокінів, які також беруть участь у розвитку якісних змін ЛПВЩ, зумовлює пригнічення атеропротекторної функції, у т. ч. захист від окиснювальної модифікації ЛППЩ. Підвищений рівень ТГ також пов’язаний з активацією каскаду коагуляційних процесів і пригніченням фібринолізу; зокрема, постпрандіальні ліпопротеїни, збагачені ТГ, активують фактор VII, який є чинником ризику ХКС. Окрім того, зміни концентрації інгібітора активатора плазміногену‑1 (ІАП‑1), що є одним із чинників ризику ГКС, позитивно корелюють з рівнем ЛПДНЩ, збагачених ТГ; зокрема, ЛПДНЩ збільшують продукцію і секрецію ІАП‑1 шляхом стимулювання експресії генів і синтезу білків ІАП‑1 у судинах.
Основні ланки патогенезу діабетичної ДЛП продемонстровано на рисунку 1 (Mooradian A. D., 2019).

Рис. 1. Патогенез діабетичної ДЛП (Mooradian A. D., 2019)
Примітки: ApoA‑1 – аполіпопротеїн A‑1; ApoB – аполіпопротеїн B; ВЖК – вільні жирні кислоти; БПЕХС – білок, який переносить ЕХ; ЕХ – ефіри ХС; ЛПЛ – ліпопротеїнліпаза; мщЛПНЩ – малі щільні часточки ЛПНЩ; ПЛ – печінкова ліпаза; ТГ – тригліцериди; TNFα – фактор некрозу пухлини α.
ІР ініціює характерну тріаду – збільшення концентрації ТГ, зменшення вмісту ХС ЛПВЩ і збільшення рівня мщЛПНЩ. Якщо концентрація ЛПДНЩ (основних транспортерів ТГ) висока, БПЕХС сприяє перенесенню ЕХ ЛПНЩ або ЕХ ЛПВЩ в обмін на ТГ. Збагачені ТГ ЛПВЩ або ЛПНЩ можуть піддаватися гідролізу ПЛ або ЛПЛ.
Основні шляхи зниження ризику приєднання та/або прогресування ДЛП при ЦД 2 типу
Одними з основних шляхів зниження ризику приєднання та/або прогресування ДЛП при ЦД 2 типу є дієта, фізична активність, корекція специфічних метаболічних порушень (гіперглікемії, порушень ліпідного обміну, симптоматична терапія супутніх захворювань і синдромів). Система фармакологічного впливу при ДЛП передбачає використання статинів, інгібіторів абсорбції ХС із кишечнику (езетиміб), інгібіторів пропротеїн конвертази субтилізин-кексинового типу 9 (РCSK9), похідних фіброєвої кислоти, нікотинової кислоти (ніацину), секвестрантів жовчних кислот (аніонообмінних смол), комбінованих препаратів, інгібітора мікросомального білка-переносника ТГ, антисмислового олігонуклеотиду, який запобігає синтезу apoВ, довголанцюгових ω3 поліненасичених жирних кислот.
Фармакологічні підходи до профілактики та лікування ДЛП
Кардіопротекторні ефекти гіполіпідемічних препаратів насамперед пояснюються їхнім впливом на метаболізм ліпідів крові. Однак гіполіпідемічні препарати також модулюють синтез і секрецію адипокінів, впливають на енергетичний гомеостаз, обмін речовин та функцію серцево-судинної (СС) системи й процеси запального каскаду в судинах, зокрема, сприяють покращенню функціонального стану ендотелію, зниженню ОС, адгезії тромбоцитів, збільшенню стабільності атеросклеротичних бляшок. Повідомляється, що зниження рівня ХС ЛПНЩ на 1,0 ммоль/л відповідає зниженню ризику ССЗ на приблизно 22%, збільшення вмісту ХС ЛПВЩ на 0,1 ммоль/л знижує ризик на приблизно 15%, тоді як відомості про ТГ остаточно не з’ясовані. Отже, першочерговим завданням за ЦД є зниження концентрації ХС ЛПНЩ (чутливішого предиктора ризику ССЗ, ніж гіперТГ або зміни вмісту ХС ЛПВЩ).
Статини
Використання інгібіторів 3-гідрокси‑3-метилглутарил-коферменту А (ГМГ-КоА)-редуктази – ключового ферменту синтезу ХС, активаторів рецепторів ЛПНЩ у печінці вважають первинною ланкою у фармакологічній стратегії лікування атерогенної ДЛП, що базується на переконливих результатах багаточисленних клінічних випробувань, зокрема, позитивного впливу на концентрацію ХС ЛПНЩ. Залежно від дози статини сприяють зниженню рівня ХС ЛПНЩ на 20-55%, спричиняють м’яке зниження ТГ (7-30%) і незначне підвищення ХС ЛПВЩ (5-10%). Зокрема, продемонстровано, що призначення симвастатину супроводжується зниженням рівня ЗХС у крові хворих на ЦД із ХКС або ГКС в анамнезі, зменшенням ризику основних коронарних подій на 55% порівняно із плацебо; використання аторвастатину в дозі 10 мг/добу сприяє зменшенню рівня ХС ЛПНЩ у крові хворих на ЦД 2 типу приблизно на 40%, зниженню відносного ризику ССЗ – на 37%, інсульту – на 48%; в іншому дослідженні встановлено, що аторвастатин (10 мг/добу) зменшує ризик ССЗ на понад 20%. Однак повідомляється, що статини (аторвастатин) не сприяють ефективності гіполіпідемічного лікування, профілактиці ХКС, зниженню ризику ССЗ у хворих на ЦД із ДЛП. Водночас використання симвастатину (80 мг/добу) в хворих із ГКС на приблизно 30% дозволяє зменшити рівень ХС ЛПНЩ, що супроводжується значним зниженням смертності. Повідомляється, що високі дози статинів (80 мг/добу аторвастатину чи 40 мг/добу симвастатину) безпечні та не пов’язані зі значним збільшенням побічних ефектів. Результати численних досліджень свідчать про ефективність статинів щодо профілактики захворювань СС-системи та зниження СС-смертності в пацієнтів із ЦД. Згідно з метааналізом, у 18 686 пацієнтів із ЦД зниження вмісту ХС ЛПНЩ на 1,0 ммоль/л (40 мг/дл) за прийому статинів асоційоване зі зменшенням загальної смертності на 9% і частотою виникнення несприятливих СС-подій. Схожі сприятливі ефекти спостерігалися при ЦД 1 та 2 типу.
Основні представники інгібіторів ГМГ-КоА-редуктази наведено в таблиці 1 (згідно із клінічними рекомендаціями Європейського товариства кардіологів (ESC), 2020).

Плейотропні ефекти статинів
Важливим аспектом є плейотропні ефекти інгібіторів ГМГ-КоА-редуктази; зокрема, гіполіпідемічні ефекти статинів супроводжуються також покращенням функції ендотелію, підвищенням стабільності атеросклеротичних бляшок, зниженням ОС і запалення, а також функціонального стану тромбоцитів. Імовірно, плейотропні ефекти статинів можуть віддзеркалювати зміни мембрани ХС, щільності ліпопротеїнів, а також динаміку їхніх взаємозв’язків із функціональними та структурними змінами клітин. Метааналіз 13 клінічних досліджень ефективності та безпеки призначення статинів 91 140 пацієнтам із ДЛП виявив, що терапія статинами пов’язана з небезпекою розвитку (в низці випадків) ЦД, однак в абсолютному значенні та порівняно з відсотком зниження ССЗ ризик виявився низьким.
Основні ефекти статинів на судинну стінку наведено в таблиці 2 (згідно зі спільними клінічними рекомендаціями ESC / Європейського товариства з атеросклерозу, EAS).
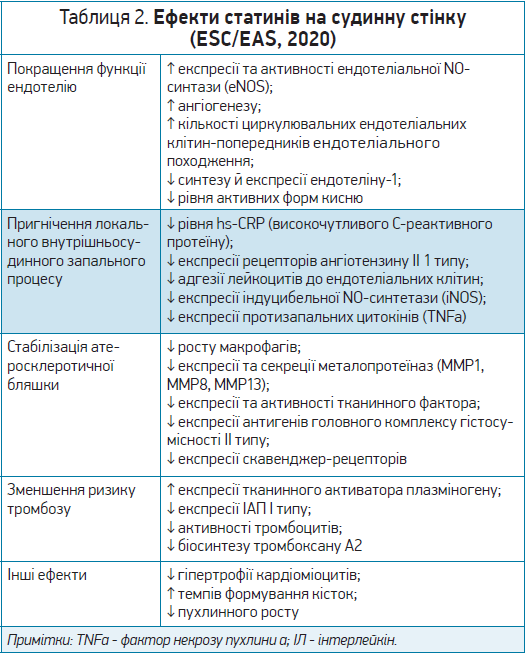
Однак необхідно зауважити, що використання інгібіторів ГМГ-КоА-редуктази асоційоване з розвитком нових випадків ЦД: зниження рівня ХС ЛПНЩ за прийому статинів на кожні 40 мг/дл супроводжується збільшенням імовірності розвитку ЦД на 10%.
Діабетогенна дія статинів – механізми та клінічні наслідки
Управління з контролю за якістю продуктів харчування та лікарських засобів США (FDA) повідомляє про можливий ризик погіршення глікемічного контролю в пацієнтів із ЦД 2 типу, а також небезпеку нових випадків ЦД 2 типу на тлі терапії статинами. Однак результати дослідження PROVE-IT TIMI 22 продемонстрували, що деякий дисбаланс глікемічного контролю в хворих на ЦД 2 типу на тлі застосування інгібіторів ГМГ-КоА-редуктази компенсується значним позитивним ефектом, що сприяє запобіганню фатальних СС-подій. Метааналіз 13 рандомізованих контрольованих досліджень (РКД) довів, що терапія статинами асоціювалася з підвищенням ризику ЦД 2 типу на 9%, зокрема, діагностуванням уперше виявленого випадку ЦД 2 типу серед 255 пацієнтів, котрі отримували лікування інгібіторами ГМГ-КоА-редуктази протягом 4 років. Однак абсолютний ризик становив лише 1 випадок на 1000 пацієнто-років лікування. Що стосується нових випадків розвитку ЦД, то FDA керується результатами дослідження JUPITER (The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin), в якому використання розувастатину як засобу первинної профілактики (порівняно із плацебо) виявило збільшення відносного ризику розвитку нових випадків ЦД 2 типу на 27%.
Інший метааналіз результатів 5 РКД (приблизно 40 тис. пацієнтів) продемонстрував, що ризик розвитку ЦД у хворих із ХКС, які нещодавно перенесли ГКС і отримували терапію статинами, становив приблизно 12%. Результати дослідження CARDS довели значне зниження ризику розвитку несприятливих СС-подій у хворих на ЦД 2 типу на тлі застосування лише 10 мг аторвастатину, однак це супроводжувалося статистично значимим погіршенням глікемічного контролю, зокрема, підвищенням рівня глікованого гемоглобіну (HbA1c) у середньому на 0,14%. Докази, що існують наразі, здебільшого базуються на аналізі post hoc (РКД або результатах метааналізу).
Ймовірні механізми розвитку статин‑індукованого ЦД 2 типу
Механізми, за допомогою яких статини можуть сприяти розвитку ЦД 2 типу, не повністю з’ясовані, але до цих процесів можуть бути залучені як цільові, так і позацільові ефекти (вплив на мевалонатний шлях, що супроводжується пригніченням декількох шляхів клітинного біосинтезу). Довготривале лікування статинами активує процеси глюконеогенезу, посилюючи експресію генів ключових ферментів, які збільшують продукцію глюкози в печінці. Крім того, інгібітори ГМГ-КоА-редуктази можуть порушувати сигнальні шляхи інсуліну, а також функціонування транспортера глюкози типу 4 (GLUT‑4), який відповідає за поглинання глюкози периферичними клітинами. Статини здатні зумовлювати зміни в циркулювальних ВЖК; гормонах, зокрема, адипонектині й лептині; функціонально-структурному стані β-клітин; дозріванні / диференціюванні адипоцитів. Додаткові механізми, зокрема, епігенетична регуляція, опосередкована специфічними мікроРНК (класом коротких некодувальних молекул РНК, що беруть участь у регуляції трансляції та деградації РНК), також задіяна в процесах зменшення секреції інсуліну.
Інгібітори ГМГ-КоА-редуктази та β-клітини підшлункової залози
Ймовірно, існує декілька механізмів приєднання та утримання гіперглікемії на тлі терапії статинами. Вважається, що реалізація ефектів інгібіторів ГМГ-КоА-редуктази може здійснюватися на рівні β-клітин підшлункової залози, тобто порушення синтезу та секреції інсуліну, а також за рахунок розвитку ІР. Інгібувальна дія статинів на білок-носій глюкози GLUT‑2 (основний носій глюкози між печінкою та кров’ю) уповільнює надходження глюкози до β-клітин підшлункової залози, отже, спричиняє зниження секреції інсуліну. Надмірне надходження ЛПНЩ до β-клітини гальмує активність глюкокінази, яка бере участь у фосфорилюванні глюкози.
Ймовірний важливий механізм діабетогенного впливу статинів – пригнічення активності ГМГ-КоА-редуктази. Зокрема, генетичні варіанти ГМГ-КоА-редуктази та лікування статинами пов’язані з надмірною масою тіла й більш високим ризиком ЦД 2 типу. Отже, ці ефекти, ймовірно, – результат пригнічення ГМГ-КоА-редуктази. Крім того, в деяких незалежних дослідженнях повідомлялося, що низькі рівні ХС ЛПНЩ асоціюються з підвищеною небезпекою приєднання ЦД. Водночас збільшення концентрації ХС ЛПНЩ у крові поєднується з нижчим ризиком ЦД, що продемонстровано за допомогою тесту, відомого як SNP-аналіз («секвенування генома бідняка», генів, пов’язаних з ліпідним обміном).
Секреція інсуліну в β-клітинах підшлункової залози ініціюється глюкозо-індукованим надходженням Ca2+, що контролюється потенціал-залежними Ca2+-каналами. Внутрішньоклітинний гомеостаз Ca2+ чітко регулюється, забезпечує належну секрецію інсуліну та підтримує фізіологічну цілісність β-клітин. Поглинання глюкози активує гліколіз у β-клітинах, отже, підвищує рівень співвідношення ATФ/AДФ-Фн, що сприяє закриттю АТФ-залежних калієвих каналів (KATФ-каналів) та деполяризації плазматичної мембрани з подальшою активацією потенціал-залежних Ca2+-каналів, надходженням позаклітинного Ca2+ і екзоцитозом інсуліну.
Взаємозв’язок між статин-індукованим пригніченням синтезу ХС і порушенням активності Са2+-каналів L-типу залишається нез’ясованим. In vitro симвастатин може безпосередньо інгібувати Са2+-канали L-типу в β-клітинах острівців підшлункової залози. Зокрема, симвастатин гальмує активність Са2+-каналів L-типу, отже, ймовірно, існує пряма взаємодія між статином і Са2+-каналом L-типу. Правастатин не має здатності пригнічувати Са2+-канали L-типу, ймовірно, внаслідок його ліпофільності. Крім того, тривале статин-опосередковане зниження рівня ХС може зумовити неправильне сортування мембранних білків, зв’язаних з ліпідним бішаром, або конформаційні зміни субодиниць Са2+-каналів. Імовірно, інгібітори ГМГ-КоА-редуктази здатні знижувати мембранний потенціал, пригнічувати активність мітохондріального комплексу II, що зумовлює ОС. Ці нецільові ефекти інгібіторів ГМГ-КоА-редуктази підтверджені експериментально; зокрема, продемонстровано, що симвастатин змінює функцію β-клітин щонайменше через два механізми: шляхом прямого інгібування KATФ-каналів; втручання в мітохондріальне дихання – внаслідок зниження рівня цитозольного АТФ і пригнічення активності Са2+-каналів L-типу.
NLRP3 – член підродини NLRP, що належить до сімейства Nod-подібних рецепторів (NLR); має ключову роль у запальних процесах у тканинах з надмірним умістом ліпідів. NLRP активує інфламасомний комплекс, який бере участь у дозріванні проінтерлейкіну (ІЛ)-1β до IЛ‑1β. Потенційними ефекторами, які можуть активувати NLRP3 при метаболічних розладах, є ВЖК і гіперглікемія. Стимулювання цитозольного білка NLRP3 (кріопірину) активує каспазу‑1 – фермент, за участю якого утворюється активна форма ІЛ‑1β, при цьому залучаються різні типи клітин-ефекторів запалення, які запускають каскад клітинних імунних реакцій, зокрема міграцію нейтрофілів до вогнища запалення й виділення зрілих форм прозапальних білків. Окрім того, порушення регуляції інфламасоми NLRP3, регуляції IЛ‑1β спричиняє зниження активності серин-треонінових кіназ (протеїнкіназ В, Akt), що беруть участь в окисному фосфорилюванні рецептора інсуліну (IR) та його субстрату (IRS). Наслідком цих патофізіологічних процесів є розвиток ІР позапечінкових клітин (переважно адипоцитів).
Основні чинники, що можуть сприяти діабетогенному впливу статинів, продемонстровано на рисунку 2 (Galicia Garcia et. al., 2020).

Рис. 2. Провідні механізми розвитку статин-індукованого ЦД 2 типу (Galicia Garcia et. al., 2020)
Примітки: GLUT‑4 – глюкозний транспортер 4 типу; GLUT‑2 – глюкозний транспортер 2 типу; мікроРНК – невелика некодувальна молекула РНК.
Статин-індукована ІР
Інгібітори ГМГ-КоА-редуктази здатні впливати на глікемічний контроль за рахунок зменшення синтезу проміжних метаболітів мевалонової кислоти, як-от ізопреноїди, фарнезилпірофосфат, геранілгеранілпірофосфат і коензим Q10 (убіхінон, CoQ10). Пряме пригнічення мевалонатного шляху статинами знижує внутрішньоклітинну концентрацію ізопреноїдів, необхідних для посттрансляційної модифікації G-білків, що важливо для екзоцитозу гранул інсуліну. Порушення синтезу ізопреноїдів спричиняє зменшення експресії GLUT‑4 та зниження надходження глюкози до адипоцитів, у яких глюкоза перетворюється на жирні кислоти (ЖК) і зберігається у вигляді ТГ. ХС-незалежні ефекти статинів можна пояснити тим, що інгібітори ГМГ-КоА-редуктази не лише регулюють синтез ХС у печінці, а й забезпечують клітини організму ізопреноїдами. Ізопреноїди, зокрема фарнезилпірофосфат і геранілгеранілпірофосфат, – це коротколанцюгові ЖК, пов’язані із сімейством клітинних сигнальних білків, «малих» G-білків (білків, що зв’язують гуанілові нуклеотиди) і належать до суперродини Ras-малих ГТФаз – Ras, Rho, Arf, G-білок Rab і Ran. Ізопреноїдна група необхідна для функціонування Ras-малих ГТФаз; деякі з них є молекулярними «перемикачами», а інші беруть участь у процесах внутрішньоклітинного мембранного транспорту. Отже, позитивні ефекти терапії інгібіторами ГМГ-КоА-редуктази обґрунтовують стимулювальну роль цих сигнальних молекул щодо клітинної проліферації, ОС, а також їхній пригнічувальний вплив на ендотеліальний синтез оксиду азоту.
Адипоцити
Нещодавно продемонстровано, що використання інгібіторів ГМГ-КоА-редуктази погіршує процес передачі сигналу інсуліну до адипоцитів, включаючи IR-білок, який кодується геном INSR. Результати численних досліджень довели, що аторвастатин і ловастатин знижують експресію GLUT‑4 на плазматичній мембрані адипоцитів лінії 3T3L1. Схожі зміни при застосуванні аторвастатину описані в білій жировій тканині в діабетичних мишей лінії NSY, які чинять порушення толерантності до глюкози (ПТГ).
Статини також порушують процеси утворення кавеол, мікродоменів плазматичної мембрани, на яких GLUT‑4 (після інсулін-стимульованої транслокації) закріплюється. Припускається, що кавеолін‑1 (Cav‑1), діючи як молекулярний шаперон, стабілізує INSR, а це необхідно для адекватної передачі сигналу інсуліну в адипоцитах. Відомо, що набуття кавеолами характерної форми залежить від вмісту ХС. Динаміка кавеол жорстко регулюється кавеоліном і кавіновими білками, тому зменшення концентрації ХС може змінити цю регуляцію. Несприятливі ефекти інгібіторів ГМГ-КоА-редуктази в кавеолах, імовірно, частково опосередковані стехіометричним зв’язуванням Cav‑1 із ХС і кавінами, що демонструє суттєву залежність структури кавеол від ХС. Статин-індуковане зниження ХС зумовлює протеасомну деградацію Cav‑2 і переміщення Cav‑1 до цитозолю, що супроводжується змінами структури кавеол. Окрім того, статин-опосередковане порушення формування характерної форми кавеол, імовірно, зменшує секрецію високомолекулярних олігомерних форм адипонектину, що знижує чутливість до інсуліну. Інгібітори ГМГ-КоА-редуктази також впливають на процеси диференціації преадипоцитів до адипоцитів.
Скелетні м’язи
GLUT‑4 опосередковує транспорт глюкози до клітин скелетних м’язів, що є ключовим чинником контролю рівня цукру в крові. Зв’язування інсуліну з INSR зумовлює активацію Akt і транслокацію везикул, які містять GLUT‑4, на плазматичну мембрану, полегшуючи в такий спосіб транспорт глюкози. Результати досліджень in vivo і in vitro свідчать про патофізіологічні зміни в скелетних м’язах, які можуть сприяти розвитку статин-індукованого ЦД 2 типу. Деякі з цих механізмів включають статин-опосередковане інгібування інсулін-стимульованого поглинання глюкози, порушення внутрішньоклітинної передачі сигналів INSR і внутрішньоклітинного сигнального шляху, центральними компонентами якого є PI3K, Akt та mTOR (PI3K/AKT/mTOR), або надмірне накопичення ВЖК у скелетних м’язах, отже, пригнічення активності ГМГ-КоА-редуктази.
Зниження експресії GLUT‑4 в культивованих міотрубках L6 після інкубації з симвастатином свідчить на користь статин-індукованої ІР у скелетних м’язах. Окрім того, аторвастатин у культивованих міотрубках C2C12 (клітинній лінії мишей) зменшує транслокацію GLUT‑4 до плазматичної мембрани та не впливає на загальну експресію білка GLUT‑4. Отже, симвастатин- або аторвастатин-асоційовані порушення транспорту глюкози в міотрубки можуть свідчити про те, що зміни внутрішньоклітинної сигналізації INSR-шляху також мають важливу роль у розвитку ІР.
Послідовність подій, котрі спричиняють симвастатин-індуковане зниження поглинання глюкози, розпочинається з порушення фосфорилювання INSR, зокрема β-ланцюга. Це супроводжується недостатнім фосфорилюванням Akt, якій, щоб стати активною, необхідне фосфорилювання за залишками Тре308 (через сигнальний шлях інсуліну) і Сер473 (за допомогою mTORC2). Симвастатин унаслідок дисрегуляції фосфорилювання mTOR (одного з компонентів mTORC2) значно погіршує фосфорилювання Akt за залишками Сер473.
Зменшення внутрішньоклітинної концентрації ХС також вважається одним із провідних механізмів порушення транслокації GLUT‑4. Симвастатин-індуковане блокування інгібіторів ГМГ-КоА-редуктази може спричинити акумулювання ацетил-КоА (попередника синтезу ЖК) і сприяти їхньому внутрішньоклітинному накопиченню. Надмірне накопичення ВЖК у скелетних м’язах супроводжується зменшенням транслокації GLUT, пригніченням поглинання глюкози, ймовірним розвитком ІР.
Печінка
Порушення чутливості печінки до інсуліну швидко відображається на гомеостазі глюкози та рівнях ТГ. Продемонстровано, що лікування інгібіторами ГМГ-КоА-редуктази пов’язане з погіршенням глікемічного контролю в печінці. Механізми, які, ймовірно, пов’язані з впливом статинів на метаболізм глюкози в печінці:
- терапія статинами пов’язана з незначним підвищенням рівня глюкози в крові натще;
- інгібітори ГМГ-КоА-редуктази здатні стимулювати ендогенне вироблення глюкози шляхом активації фосфоенолпіруваткарбоксикінази (PEPCK) і глюкозо‑6-фосфатази – основних ферментів, що обмежують швидкість глюконеогенезу в гепатоцитах людини;
- активація процесів глюконеогенезу в печінці, як відомо, сприяє характерній для ІР і ЦД 2 типу гіперглікемії;
- накопичення ВЖК у гепатоцитах може супроводжуватися розвитком ЦД 2 типу.
Статини та мікроРНК
МікроРНК – це невеликі (22 нуклеотиди) некодувальні регуляторні РНК, які функціонують як посттранскрипційні регулятори експресії генів. МікроРНК залучені до багатьох біологічних процесів – експресії інсуліну, адаптації скелетних м’язів до підвищення рівня глюкози, чутливості до інсуліну та глюкозо-стимульованої секреції інсуліну. Встановлено, що лікування статинами впливає на експресію декількох мікроРНК, що мають центральну роль у регуляції метаболізму ліпідів і глюкози, а також пов’язані з розвитком ЦД 2 типу.
МікроРНК‑33 також негативно модулює експресію IRS2, впливаючи в такий спосіб на передачу сигналу інсуліну. МікроРНК‑33а та мікроРНК‑33b – це інтронічні міРНК, які знаходяться в межах генів білка, що зв’язує регуляторний елемент стеролу 2 (SREBP2) і SREBP1 відповідно. МікроРНК‑33а і мікроРНК‑33b беруть участь у регуляції метаболізму ЖК, ліпідів і глюкози, а також залучені до регуляції метаболічних шляхів, які впливають на основні чинники ризику ІР. МікроРНК‑33a та мікроРНК‑33b (разом зі SREBP2, SREBP1) модулюють внутрішньоклітинний гомеостаз ХС і ЖК. МікроРНК‑33a/b регулюють гомеостаз ХС/ліпідів шляхом зв’язування в 3’UTR генів (3’-кінцевих ділянок, котрі не транслюються, особливих ділянок мікроРНК, які не слугують матрицею для синтезу білків), що беруть участь у транспорті ХС, як-от транспортери АТФ-зв’язувальної касети 1 (ABCА1) та ABCG1, і посилюють або пригнічують його експресію. Крім того, продемонстровано, що іn vivo мікроРНК‑33a пригнічує експресію ABCА1 та ABCG1. Отже, мікроРНК‑33a – важливий регулятор АBCA1 та ABCG1, а рівні їхньої експресії у β-клітинах є зворотно пропорційними. ABCA1 має важливу роль у запобіганні накопиченню ХС у макрофагах, зокрема, опосередковує транспорт ХС із периферичних тканин до Apo‑1, а також у зворотному транспортному шляху ХС. Експресія miR‑33 спричиняє зниження рівня ABCA1, що зумовлює зменшення надходження ХС до ApoА‑1. Пригнічення функції miR‑33 спричиняє зростання рівня ABCA1 і збільшення транспортування ХС до ApoА‑1. МікроРНК‑33a-опосередковане зниження регуляції ABCA1 також здатне змінити гомеостаз ХС і порушити секрецію інсуліну, що зумовлює дисфункцію β-клітин. Установлено, що симвастатин та аторвастатин індукують експресію мікроРНК‑33a в печінці, що свідчить про зв’язок між зниженою секрецією інсуліну й розвитком статин-індукованого ЦД 2 типу.
Родина мікроРНК‑27 (мікроРНК‑27a і мікроРНК‑27b) – ключові регулятори обміну ХС і гомеостазу ліпідів. МікроРНК‑27a безпосередньо пригнічує високоафінний рецептор ЛПНЩ (ЛПНЩ-рецептор) РНК і рівні білка, зв’язуючись з 3’UTR ЛПНЩ-рецептора мікроРНК. Окрім того, мікроРНК‑27a опосередковано (завдяки посиленню регуляції інгібіторів PCSK9) зменшує експресію ЛПНЩ-рецептора.
Повідомляється, що в клітинах гепатоцелюлярної карциноми людини активність мікроРНК‑27a і мікроРНК‑27b дозозалежно регулюється симвастатином. Окрім того, на тлі ЦД 2 типу за хронічної гіперглікемії спостерігається дисрегуляція мікроРНК‑27a в жировій тканині та в адипоцитах лінії 3T3-L1.
Продемонстровано, що вплив інгібіторів ГМГ-КоА-редуктази на процеси утворення глюкози печінкою опосередковується через модуляцію експресії ферментів глюконеогенезу, активацію кластера мікроРНК‑183/96/182. Зокрема, інкубація гепатоцитів з аторвастатином, симвастатином або правастатином посилює експресію PEPCK і глюкозо‑6-фосфатази. Ефекти інгібіторів ГМГ-КоА-редуктази включають мікроРНК‑183/96/182-опосередковане зниження регуляції фактора транскрипції 7, подібного фактора 2 (TCF7L2), який модулює печінковий і периферичний метаболізм глюкози та контроль глікемії. TCF7L2 зменшує активність процесів глюконеогенезу в печінці, ймовірно, інгібуванням транскрипційної активності позитивних регуляторів PEPCK і глюкозо‑6-фосфатази. Результати низки досліджень свідчать, що в пацієнтів, які отримують тривале лікування статинами, спостерігається підвищена експресія кластера мікроРНК, а це спричиняє стійку активацію глюконеогенезу, отже, сприяє розвитку ЦД 2 типу.
Результати експериментальних робіт in vivo та in vitro, клінічних досліджень підтверджують діабетогенну дію інгібіторів ГМГ-КоА-редуктази. Хоча безліч питань на сьогодні нез’ясовані, фактичні дані свідчать, що статини збільшують ризик розвитку ЦД 2 типу; вплив деяких інгібіторів ГМГ-КоА-редуктази є вищим за інших (наприклад, симвастатин, розувастатин, аторвастатин порівняно із правастатином).
Основні положення при оцінці ризику розвитку статин-індукованого ЦД
Під час оцінки ризиків розвитку ЦД слід ураховувати такі положення:
- в пацієнтів з АССЗ, які отримували статини, ризик приєднання ЦД є на щабель нижчим, ніж небезпека розвитку серйозних несприятливих СС-подій;
- предіабет діагностований у багатьох хворих, у яких спостерігалася маніфестація ЦД (надмірна маса тіла, ІР, генетична схильність тощо);
- застосування статинів могло прискорити появу ЦД на декілька місяців, але зазвичай не спричиняє розвитку ЦД у нетолерантних до нього людей;
- ризик ЦД пов’язаний із застосуванням високоінтенсивної терапії статинами чи високих доз препаратів інгібіторів ГМГ-КоА-редуктази;
- застосування статинів при маніфестації ЦД надає кількісно більші переваги, оскільки ризик розвитку АССЗ у таких хворих також збільшується.
Отже, в більшості клінічних рекомендацій зазначено, що сукупні переваги статинів переважають занепокоєння, пов’язані з можливістю розвитку ЦД.
Ключові положення, пов’язані з діабетогенною дією статинів:
- метааналіз даних клінічних випробувань виявив збільшення ризику вперше діагностованого ЦД, пов’язаного з терапією статинами, на 10-12%, причому ризик зростав при використанні інтенсивних схем лікування;
- результати менделівських рандомізованих випробувань свідчать, що вплив інгібіторів ГМГ-КоА-редуктази на метаболізм глюкози сприяє зменшенню ефективності основної мети терапії статинами – пригнічення активності інгібіторів ГМГ-КоА-редуктази;
- дані in vitro та in vivo свідчать про те, що статини зменшують синтез продуктів мевалонатного шляху та збільшують навантаження клітин ХС, що зумовлює порушення функції β-клітин і зниження чутливості до інсуліну;
- переваги статинів у зменшенні ССЗ значно перевищують негативні наслідки ризику приєднання ЦД, отже, застосування інгібіторів ГМГ-КоА-редуктази має залежати від індивідуального ризику ЦД;
- застосування статинів до встановлення діагнозу ЦД не збільшує поширеності мікросудинних захворювань; крім того, вплив інгібіторів ГМГ-КоА-редуктази на глікемічний контроль є незначним у пацієнтів із ЦД;
- пацієнтам, які приймають статини (особливо хворим із чинниками ризику розвитку ЦД), необхідно контролювати можливість розвитку дисглікемії, а також надавати відповідні поради щодо дієти і способу життя.
Основні положення, пов’язані з використанням статинів у пацієнтів із високим ризиком розвитку ЦД
Основні положення, пов’язані з використанням статинів у пацієнтів із високим ризиком розвитку ЦД:
- статини – обов’язковий компонент багатофакторної профілактики СС-ускладнень у хворих на ЦД;
- користь від застосування статинів як засобів первинної та вторинної профілактики в пацієнтів дуже високого, високого і помірного ризику значно перевершує ризик виявлення нових випадків ЦД;
- механізми розвитку порушень вуглеводного обміну на тлі ліпідознижувальної терапії статинами є складними та наразі недостатньо вивченими. Ймовірно, існує відмінність за частотою розвитку ЦД у пацієнтів, які отримують різні лікарські препарати із групи інгібіторів ГМГ-КоА-редуктази;
- ризик розвитку ЦД закономірно пов’язаний із чинниками ризику захворювання в конкретного пацієнта та в більшості випадків спостерігається в хворих із ПТГ.
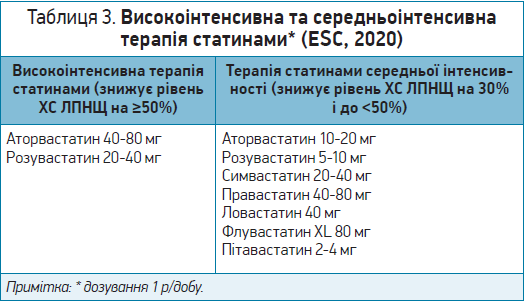
Особливості високоінтенсивної та середньоінтенсивної терапії статинами наведено в таблиці 3.
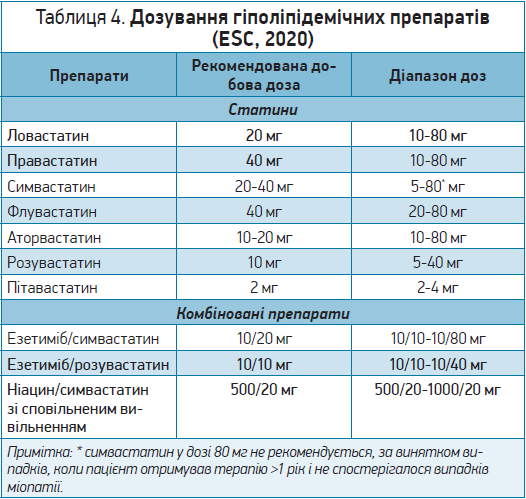
Особливості дозування гіполіпідемічних препаратів наведено в таблиці 4.
У таблиці 5 наведено рекомендації щодо особливостей медикаментозної терапії ДЛП у хворих на ЦД.
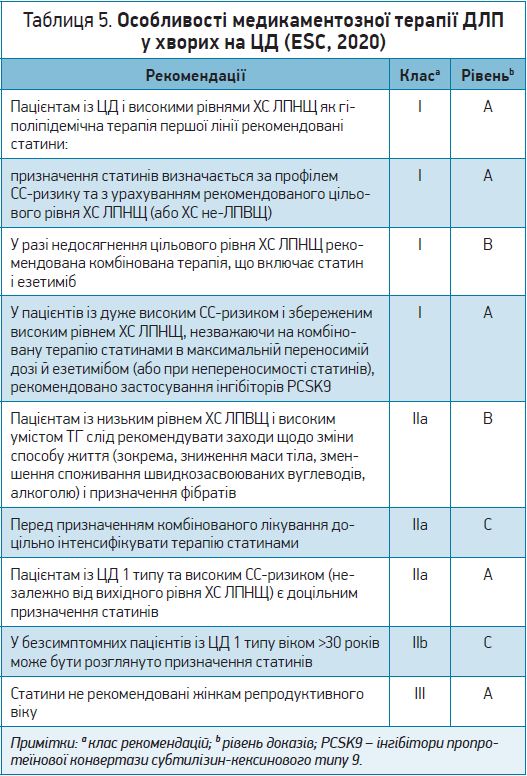
Провідні експерти ЕАS рекомендують при вирішенні питання про можливість використання статинів у пацієнтів із групи ризику ЦД зважувати співвідношення ризику та вигоди. Очевидно, що користь від застосування статинів у хворих із підвищеним ризиком ССЗ набагато перевищує наявний незначний абсолютний ризик розвитку ЦД 2 типу. Навіть якщо в пацієнта розвивається ЦД, ризики, пов’язані з ССЗ, є набагато більшими, ніж небезпека приєднання ЦД. Перед початком терапії статинами необхідно попередньо оцінити ступінь ризику ЦД. В осіб із високим ризиком розвитку ЦД 2 типу, які отримують статини, необхідно регулярно контролювати рівні HbA1c і глюкози в крові. Ризик переходу від ПТГ до ЦД може бути знижений за рахунок зміни способу життя, а також використання цукрознижувальної терапії. Якщо в пацієнта розвивається ЦД під час лікування статинами, терапію статинами слід продовжувати та здійснювати лікування ЦД відповідно до положень національних рекомендацій. Ця позиція знайшла відображення в рекомендаціях ESC/EAS щодо лікування ДЛП (2020).
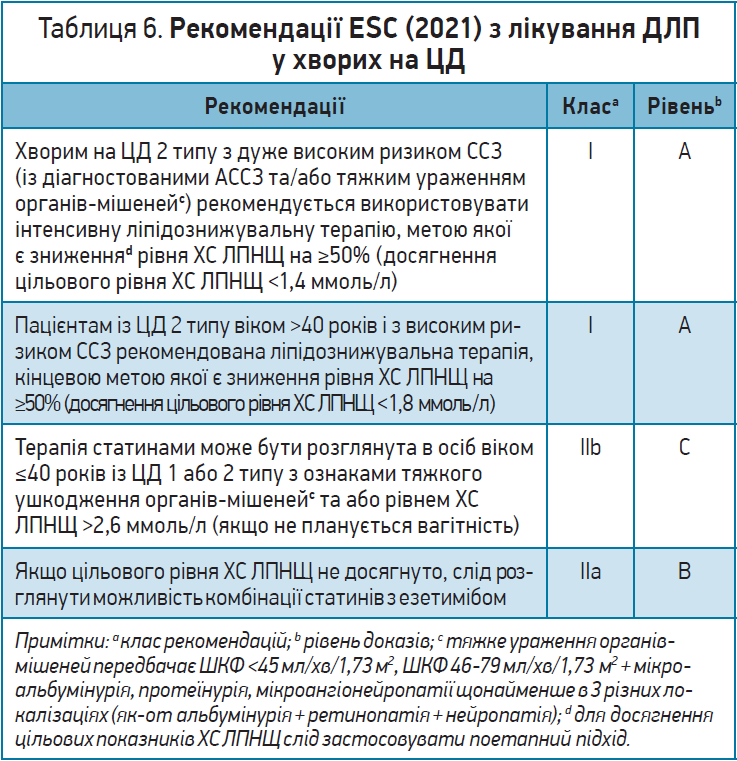
ESC (2021) пропонує (як і за профілактики ССЗ у здорових людей) поетапний підхід до контролю ліпідів залежно від ризику, очікуваної користі протягом усього життя, супутніх захворювань та переваг гіполіпідемічної терапії у конкретного пацієнта. Відповідно до нових рекомендацій з профілактики ССЗ, опублікованих ESC у 2021 р., для пацієнтів зі встановленим АССЗ і для хворих на ЦД із дуже високим ризиком (із діагностованим АССЗ чи тяжким ураженням органів-мішеней) цільове значення ХС ЛПНЩ має становити <1,4 ммоль/л (або зниження вихідного показника на ≥50%); для хворих із ЦД віком >40 років і високим ризиком – 1,8 ммоль/л (чи зниження вихідного показника на ≥50%). У вищезазначеному документі наведено рекомендації для відносно здорових людей віком >70 років із дуже високим і високим ризиком ССЗ: цим категоріям слід підтримувати вміст ХС ЛПНЩ на рівні <1,4 ммоль/л і <1,8 ммоль/л відповідно чи знижувати вихідний показник на ≥50%. У хворих на ЦД, які не досягають цільових показників ХС ЛПНЩ за допомогою статинів та/або езетимібу, можна використовувати інгібітори PCSK9 (табл. 6).
Висновки
На сьогодні на молекулярному рівні тривають дослідження, спрямовані на з’ясування статин-індукованих механізмів розвитку ЦД 2 типу. В світлі результатів численних обсерваційних досліджень установлено, що терапія інгібіторами ГМГ-КоА-редуктази впливає на приєднання ЦД 2 типу, однак сприяє зниженню приєднання та/або прогресування ССЗ. Отже, прийом статинів слід продовжувати пацієнтам із високим або дуже високим ризиком ССЗ для досягнення цільових рівнів ХС ЛПНЩ. Необхідно пам’ятати, що перед початком призначення інгібіторів ГМГ-КоА-редуктази необхідно оцінити ризик розвитку ЦД.
