


13 березня, 2024
Хвороба Гоше: нові міркування та проблеми раннього виявлення розладу
Хвороба Гоше (ХГ) є рідкісним тяжким спадковим лізосомним захворюванням накопичення, яке при відсутності своєчасної діагностики й адекватного лікування може призвести до інвалідизації та скорочення тривалості життя пацієнта. Незважаючи на низьку поширеність, ХГ має велике медико-соціальне значення через виражені клінічні прояви, які значно знижують якість життя пацієнтів, і значні фінансові витрати на лікування й реабілітацію. Рання діагностика захворювання є запорукою своєчасного призначення патогенетичної ферментнозамісної терапії (ФЗТ), спрямованої на мінімізацію розвитку незворотних ускладнень, досягнення стійкої ремісії та збереження соціальної активності і працездатності. Відповідно, величезне значення має обізнаність лікарів первинної ланки, особливо сімейних лікарів і педіатрів, щодо основних клінічних ознак ХГ для своєчасної діагностики і негайного направлення пацієнтів на консультацію до генетика задля верифікації діагнозу й початку лікування.
Лізосомальні хвороби накопичення (ЛХН) – це група спадкових захворювань, пов’язаних з порушенням функції лізосом, внутрішньоклітинних органел, що перетравлюють екзогенний матеріал або непотрібні органели клітини за допомогою ферментів. Генетично детерміноване порушення синтезу одного або декількох ферментів лізосом призводить до накопичення в них специфічного субстрату цих ферментів. Клінічна картина окремих ЛХН залежить від того, в яких органах і тканинах відбувається накопичення субстрату. Вони часто мають поліорганний характер і потребують диференційної діагностики із захворюваннями нервової системи, нирок, м’язовою дистрофією, скелетною дисплазією, гепатомегалією, спленомегалією, кардіоміопатією тощо.
Однією з найбільш поширених ЛХН є ХГ, відома також як сфінголіпідоз – розлад, спричинений дефіцитом ферменту глюкоцереброзидази (GCase). Згідно з останньою класифікацією спадкових метаболічних розладів [1], ХГ визначається як вроджена помилка метаболізму з аутосомно-рецесивним типом успадкування, яка належить до підкатегорії ЛХН [2]. Дефіцит ферменту призводить до накопичення глюкоцереброзиду в лізосомах і подальшого ураження багатьох органів. Недостатній катаболізм глюкозилцераміду і накопичення макрофагами цього субстрату призводять до вісцеральних проявів ХГ. Сфінголіпіди беруть участь у запальних та апоптотичних процесах, а глюкозилцерамід може мати прямий активуючий або посилюючий вплив на функцію макрофагів, можливо, через селективну дисрегуляцію кальцієвих каналів. Декілька індикаторів активації макрофагів були виявлені у надлишку в плазмі пацієнтів із ХГ [3, 4]. Гістологічна оцінка показала неоднакове збільшення деяких молекул у клітинах селезінки при ХГ [5]. Альтернативним механізмом є аномальне згортання мутантних білків в ендоплазматичному ретикулумі [6], що ініціює реакцію розгорнутого білка і може викликати апоптоз або запалення [7]. Існують докази, що деякі мутації при ХГ можуть призвести до аномально згорнутих чи транспортованих білків [8].
Незалежно від основного генетичного дефекту, ХГ призводить до мультисистемного розладу, що характеризується фенотиповою гетерогенністю і широким клінічним спектром. Сучасна література визначає 3 основні фенотипи:
- тип 1 (ХГ1 хронічна ненейронопатична) – ненейронопатичний варіант із переважним ураженням печінки, селезінки, кісток і гематологічної системи;
- тип 2 (ХГ2 гостра нейронопатична) – гострий нейронопатичний варіант, який виникає в ранньому дитинстві, є найтяжчою формою;
- тип 3 (ХГ3 хронічна нейронопатична) – підгострий нейронопатичний варіант, демонструє клінічний початок частіше в дитинстві або підлітковому віці [2].
Ці 3 основні фенотипи можна додатково класифікувати на підтипи залежно від віку появи симптомів, швидкості прогресування та інших ознак [9]. Найпоширенішою формою в Європі, Ізраїлі, Канаді і США є ХГ1 (94%), в інших країнах частішими є нейронопатичні форми [10, 11].
Портрет педіатричного пацієнта з ХГ
ХГ має різноманітні клінічні прояви, добре описані в літературі [14]. Симптоми ХГ збігаються з проявами деяких поширених дитячих захворювань, що призводить до діагностичних помилок і затримки встановлення правильного діагнозу (рис. 1) [15].
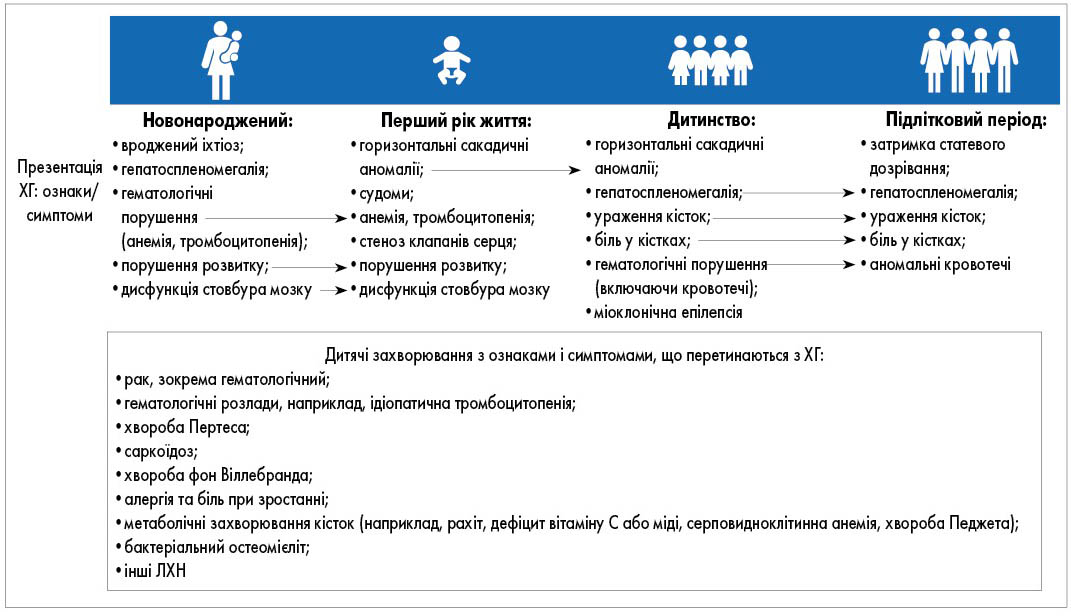 Рис. 1. Хронологія появи ознак і симптомів ХГ у дитячому віці та детальна інформація про інші дитячі захворювання, ознаки і симптоми яких перетинаються з ХГ (N.J. Weinreb et al., 2022)
Рис. 1. Хронологія появи ознак і симптомів ХГ у дитячому віці та детальна інформація про інші дитячі захворювання, ознаки і симптоми яких перетинаються з ХГ (N.J. Weinreb et al., 2022)
У дітей із ХГ1 найчастішими проявами є спленомегалія, гепатомегалія, тромбоцитопенія, носові кровотечі тощо, що значно погіршує якість життя [12, 13]. Середня тривалість життя при ХГ1 на 10 років менша за загальну популяцію. Діти з ХГ2, як правило, помирають до 2 років, тоді як при ХГ3 неврологічні симптоми розвиваються повільніше і хворі часто доживають до зрілого віку [16].
Спленомегалію та цитопенію при ХГ можна сплутати з іншими хворобами [17], зокрема, зі злоякісними гематологічними новоутвореннями. Навіть у разі низької ймовірності раку це припущення розглядають першочергово, що викликає занепокоєння у батьків. Якщо рак виключено, слід розглянути вірогідність наявності ХГ. За даними опитування, 43% пацієнтів із ХГ у США отримали попередній помилковий діагноз лейкемії, множинної мієломи або раку печінки [18, 19]. Враховуючи це, була визначена низка ознак і параметрів, найбільш характерних для ранніх стадій ХГ1 і ХГ3, щоб полегшити діагностику цих захворювань (табл.) [14]. 
Загалом усі типи ХГ можуть характеризуватися ураженням внутрішніх органів зі спленомегалією та/або гепатомегалією [21]. Інші результати можуть включати цитопенію з тромбоцитопенією, анемію, лейкопенію, ураження кісток, інфільтрацію кісткового мозку, біль у кістках з остеопенією та системні симптоми, такі як затримка росту або затримка статевого дозрівання [22].
Алгоритм діагностики ХГ
Діагностика ХГ ускладнюється не тільки гетерогенністю і неспецифічністю симптомів, а також географічною та віковою варіабельністю. Неврологічні прояви більш поширені в азіатських та арабських популяціях, ніж у переважно кавказьких (5% ХГ2/ХГ3). Спостерігаються також регіональні варіації: у Швеції 40% випадків ХГ3 діагностують на півночі. Різні симптоми зазвичай з’являються у певному віці. Диференційна діагностика, як правило, охоплює широкий спектр інфекційних, злоякісних і метаболічних захворювань. Ці неспецифічні симптоми, фенотипова неоднорідність і відсутність знань про хворобу часто призводять до затримки діагностики, а іноді й до тривалої діагностичної «одісеї» навіть у дітей з явними клінічними проявами [22].
Враховуючи гетерогенність захворювання, було запропоновано переглянути підхід до алгоритму діагностики ХГ з фокусом на походженні пацієнтів і наявності спленомегалії як ключової ознаки ХГ, оскільки вона присутня у переважної більшості пацієнтів цієї групи [23]. Згідго із сучасними даними, 87% пацієнтів із ХГ мають спленомегалію, що перевищує норму у 5 разів (середній об’єм селезінки 15,2×норма) [24]. У пацієнтів ашкеназького походження частота ХГ становить приблизно 1:800, тоді як гематологічні злоякісні новоутворення зустрічаються набагато рідше (близько 1:2 500) [25]. Тому в цій етнічній групі доцільно проводити тестування на ХГ як обстеження 1-ої лінії за наявності спленомегалії та цитопенії. Слід враховувати, що найпоширеніший генотип у цій групі часто характеризується легкою цитопенією та спленомегалією, які спочатку можуть залишитися непоміченими. Такі ознаки, як гіперферитинемія, низький рівень ліпопротеїдів високої щільності, жовчнокам’яна хвороба чи остеопороз, мають насторожити щодо можливої ХГ.
У неашкеназьких популяціях ХГ зустрічається рідше (1:40 000) порівняно зі злоякісними гематологічними новоутвореннями. Тому спочатку слід виключити рак, а потім розглядати ХГ. Під час біопсії кісткового мозку варто шукати ознаки як злоякісних новоутворень, так і клітин Гоше. Це обґрунтовує діагностичний алгоритм, стратифікований за етнічною приналежністю (рис. 2, 3).
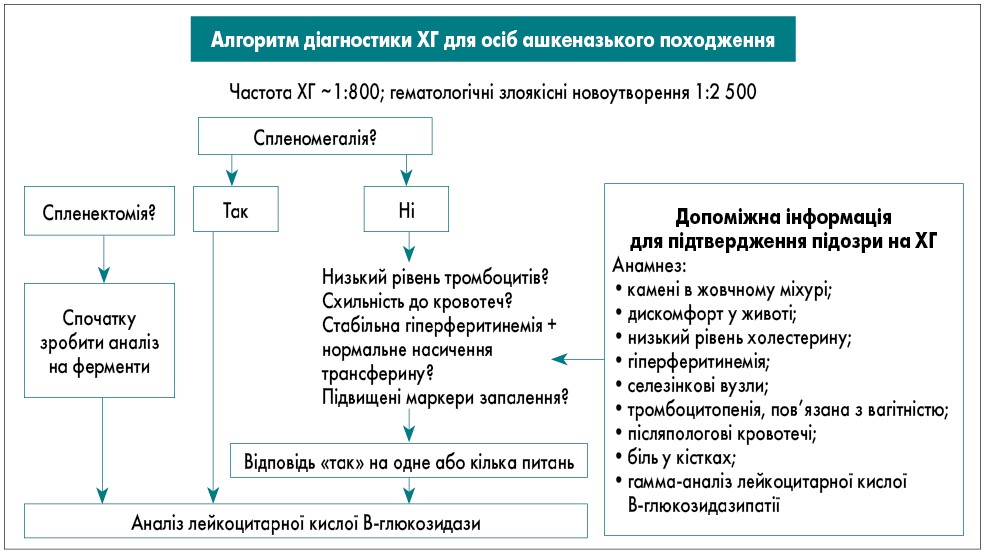 Рис. 2. Алгоритм діагностики ХГ в осіб ашкеназького походження (P.K. Mistry et al., 2011)
Рис. 2. Алгоритм діагностики ХГ в осіб ашкеназького походження (P.K. Mistry et al., 2011)
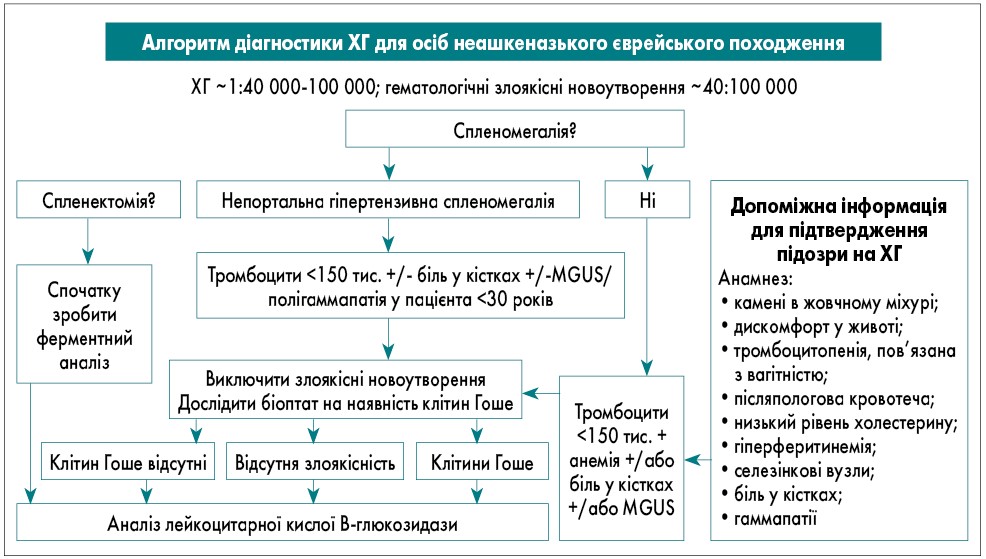 Рис. 3. Алгоритм діагностики ХГ для осіб неашкеназького єврейського походження (P.K. Mistry et al., 2011)
Рис. 3. Алгоритм діагностики ХГ для осіб неашкеназького єврейського походження (P.K. Mistry et al., 2011)
Примітки: MGUS – моноклональна гамапатія неуточненого генезу.
Діагностичним тестом для ХГ є демонстрація низької активності кислотної β-глюкозидази в лейкоцитах периферичної крові (норма 2,1-5,3 мкмоль/л/год). Дослідження проводять на лейкоцитах з використанням флуоресцентного субстрату [23].
! Проведення секвенування всієї кодуючої ділянки гена GBA1 рекомендовано пацієнтам із високою підозрою на ХГ у разі негативних результатів скринінгу на поширені мутації.
Аналіз мутацій GBA1 може надати прогностичну інформацію, хоча фенотипічна мінливість серед пацієнтів з однаковим генотипом GBA1 значна. Визначення мутації GBA1 у пробанда також полегшує сімейний скринінг для генетичного консультування, оскільки гетерозиготних носіїв неможливо надійно ідентифікувати за допомогою ферментних тестів.
Фармакологічне лікування ХГ у дітей
Раніше пацієнти з ХГ отримували переважно симптоматичне лікування, включаючи спленектомію та ортопедичні операції. Окремим пацієнтам проводили алогенну трансплантацію гемопоетичних стовбурових клітин. Спленектомія була показана за наявності масивної спленомегалії та вираженого гіперспленізму з метою корекції порушень харчування, затримки росту у дітей або усунення механічних чи судинних ускладнень [26]. Проте після спленектомії у переважної більшості хворих спостерігали розвиток кісткових ускладнень, таких як остеонекроз великих суглобів, прогресування гепатомегалії з розвитком цирозу печінки та підвищення ризику летальних випадків від септицемії [27]. З впровадженням ФЗТ показання до спленектомії практично зникли [28].
Сучасні підходи до лікування ХГ включають ФЗТ та субстрат-редукуючу терапію (СРТ). Метою лікування є запобігання інвалідизації внаслідок ускладнень, таких як масивна фіброзна спленомегалія, аваскулярний некроз, вторинний остеоартрит, компресія хребців та переломи, фіброз печінки чи легень. Окрім специфічного лікування, може знадобитися симптоматична терапія, наприклад, призначення аналгетиків при болю в кістках, препаратів кальцію та вітаміну D при остеопорозі, гемотрансфузії при вираженій анемії чи геморагічному синдромі [19].
Концепція ФЗТ була вперша описана у 1966 р., через рік після встановлення успадкованого дефіциту GCase як етіологічного фактора ХГ [29, 30]. Спроби ФЗТ при ХГ проводилися з середини 1970-х років, але були невдалими до того часу, доки маніпуляції з ферментом не призвели до відкриття внутрішніх залишків манози, що забезпечило спрямування ферменту до макрофагів [31]. ФЗТ вперше була успішно застосована в клініці у 1991 р. як терапія для пацієнтів із ХГ1 і ХГ3 [32]. Пізніше з’явився препарат іміглюцераза – рекомбінантний аналог GCase, який продукується в клітинах яєчників китайського хом’ячка. Принцип ФЗТ полягає у заміщенні дефіцитного ферменту GCase у клітинах, особливо в клітинах Гоше [33].
! Ефективність і переносимість ФЗТ пацієнтами із ХГ підтверджена результатами клінічних досліджень, даними програм фармаконагляду виробників препаратів, а також інформацією з реєстрів захворювань і лікарських засобів. Не повідомлялося про випадки смерті або незворотного ушкодження внаслідок тяжких побічних реакцій, пов’язаних із препаратом [34].
У відкритому 9-місячному дослідженні взяли участь 12 дорослих пацієнтів (≥18 років) з ХГ1, які раніше не отримували ФЗТ. Велаглюцеразу-α спочатку призначили трьом пацієнтам у зростаючих дозах (15, 30, 60 ОД/кг), іншим 9-ти – одразу по 60 ОД/кг. Вже після 3-х місяців лікування спостерігали клінічно значуще збільшення рівнів гемоглобіну і тромбоцитів, зменшення об’ємів печінки і селезінки – після 6 і 9 місяців відповідно. Після ≥12 місяців терапії велаглюцеразою-α у дозі 60 ОД/кг усім зменшили дозу до 30 ОД/кг після досягнення ≥2 із 4-х терапевтичних цілей ФЗТ на першому році. Пацієнти отримували терапію у дозах 30-60 ОД/кг (у середньому 35 ОД/кг) 1 раз на 2 тижні протягом 7 років. Продемонстрована стійка клінічна ефективність у вигляді поліпшення показників крові і зменшення розмірів органів. До 57-го місяця в усіх відмічене зниження навантаження кісткового мозку за даними МРТ, а також покращення мінеральної щільності кісткової тканини [35].
У 12-місячному подвійному сліпому рандомізованому дослідженні з паралельною групою порівняння взяли участь 25 раніше нелікованих за допомогою ФЗТ пацієнтів із ХГ віком від 2 років з анемією, тромбоцитопенією або органомегалією. Їх рандомізували до груп отримання велаглюцерази-α у дозах 45 або 60 ОД/кг внутрішньовенно 1 раз на 2 тижні. Згідно з отриманими даними, введення велаглюцерази-α у дозі 60 ОД/кг продемонструвало клінічно значуще збільшення середніх рівнів гемоглобіну (+2,4 г/дл) і кількості тромбоцитів (+50,9×109/л), а також зменшення об’ємів печінки й селезінки в середньому на 17 та 50% відповідно порівняно з вихідними даними. При дозі 45 ОД/кг також спостерігалося значуще збільшення рівнів гемоглобіну (+2,4 г/дл) і тромбоцитів (+40,9×109/л) та зменшення об’ємів печінки на 6% і селезінки на 40%.
Таким чином, ХГ є прогресуючим мультисистемним захворюванням, яке без адекватного лікування може призвести до розвитку незворотних ускладнень та інвалідизації пацієнтів. Відповідно, своєчасне виявлення цієї патології вкрай важливе для призначення етіопатогенетичної терапії до розвитку незворотних змін і для досягнення оптимального контролю симптомів захворювання. ФЗТ, зокрема, застосування велаглюцерази-α, має на меті зменшення органомегалії, корекцію цитопенії, а також потенційно може чинити позитивний вплив на стан кісткової тканини, що може сприяти профілактиці розвитку ускладнень і збереженню працездатності пацієнтів.
Література
- Wraith J.E. The clinical presentation of lysosomal storage disorders. Acta Neurol Taiwan. 2004 Sep; 13 (3): 101-6.
- Pession A., Di Rocco M., Venturelli F. et al. GAU-PED study for early diagnosis of Gaucher disease in children with splenomegaly and cytopenia. Orphanet J Rare Dis. 2023 Jun 16; 18 (1): 151. doi: 10.1186/s13023-023-02760-z.
- Gordon S., Taylor P.R. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005 Dec; 5 (12): 953-64. doi: 10.1038/nri1733.
- Brinkman J., Wijburg F.A., Hollak C.E. et al. Plasma chitotriosidase and CCL18: early biochemical surrogate markers in type B Niemann-Pick disease. J Inherit Metab Dis. 2005; 28 (1): 13-20. doi: 10.1007/s10545-005-4416-9.
- Boven L.A., van Meurs M., Boot R.G. et al. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am J Clin Pathol. 2004 Sep; 122 (3): 359-69. doi: 10.1309/BG5V-A8JR-DQH1-M7HN.
- Schroder M., Kaufman R.J. ER stress and the unfolded protein response. Mutat Res. 2005 Jan 6; 569 (1-2): 29-63. doi: 10.1016/j.mrfmmm.2004.06.056.
- Gargalovic P.S., Gharavi N.M., Clark M.J. et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006 Nov; 26 (11): 2490-6. doi: 10.1161/01.ATV.0000242903.41158.a1.
- Tessitore A., del P Martin M., Sano R. et al. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell. 2004 Sep 10; 15 (5): 753-66. doi: 10.1016/j.molcel.2004.08.029.
- Kaplan P., Baris H., De Meirleir L. et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr; 172 (4): 447-58. doi: 10.1007/s00431-012-1771-z.
- Tylki-Szymanska A., Vellodi A., El-Beshlawy A. et al. Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J Inherit Metab Dis. 2010 Aug; 33 (4): 339-46. doi: 10.1007/s10545-009-9009-6.
- Puri R.D., Kapoor S., Kishnani P.S. Diagnosis and Management of Gaucher Disease in India - Consensus Guidelines of the Gaucher Disease Task Force of the Society for Indian Academy of Medical Genetics and the Indian Academy of Pediatrics. Indian Pediatr. 2018 Feb 15; 55 (2): 143-153.
- Stirnemann J., Belmatoug N., Camou F. et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int J Mol Sci. 2017 Feb 17; 18 (2): 441. doi: 10.3390/ijms18020441.
- Remor E., Baldellou A. Health-related quality of life in children and adolescents living with Gaucher disease and their parents. Health Psychol Behav Med. 2018 Apr 12; 6 (1): 79-92. doi: 10.1080/21642850.2018.1462705.
- Mehta A., Kuter D.J., Salek S.S. et al. Presenting signs and patient co-variables in Gaucher disease: outcome of the Gaucher Earlier Diagnosis Consensus (GED-C) Delphi initiative. Internal medicine journal, 49 (5), 578-591. https://doi.org/10.1111/imj.14156.
- Roshan Lal T., Sidransky E. The Spectrum of Neurological Manifestations Associated with Gaucher Disease. Diseases. 2017 Mar 2; 5 (1): 10. doi: 10.3390/diseases5010010.
- Mistry P.K., Belmatoug N., vom Dahl S. et al. Understanding the natural history of Gaucher disease. Am J Hematol. 2015 Jul; 90 Suppl 1:S6-11. doi: 10.1002/ajh.24055.
- Nagral A. Gaucher disease. J Clin Exp Hepatol. 2014 Mar; 4 (1): 37-50. doi: 10.1016/j.jceh.2014.02.005.
Повний список літератури знаходиться в редакції.
Підготувала Анна Сочнєва
Матеріал створенний за підтримки компанії «ТАКЕДА». Містить рекламу.
© ТОВ «ТАКЕДА Україна». Всі права захищені.
«ТАКЕДА» та ![]() є зареєстрованими торговельними марками компанії Takeda Pharmaceutical Company Limited
є зареєстрованими торговельними марками компанії Takeda Pharmaceutical Company Limited
VV-MEDMAT-100538
Тематичний номер «Педіатрія» № 1 (72) 2024 р.
