


26 грудня, 2020
Орфанні захворювання в ендокринології
 Медико-соціальне значення орфанних захворювань (ОЗ) зумовлене складнощами в діагностиці й терапії, а також неблагоприємним прогнозом і високою частотою інвалідизації пацієнтів. Ухвалення Національної стратегії з профілактики, діагностики та лікування ОЗ в Україні свідчить про визнання проблеми на державному рівні та дає можливість вирішення питань своєчасного виявлення рідкісних захворювань, лікування та забезпечення пацієнтів життєво необхідними лікарськими засобами.
Медико-соціальне значення орфанних захворювань (ОЗ) зумовлене складнощами в діагностиці й терапії, а також неблагоприємним прогнозом і високою частотою інвалідизації пацієнтів. Ухвалення Національної стратегії з профілактики, діагностики та лікування ОЗ в Україні свідчить про визнання проблеми на державному рівні та дає можливість вирішення питань своєчасного виявлення рідкісних захворювань, лікування та забезпечення пацієнтів життєво необхідними лікарськими засобами.
У 1983 р. в США було прийнято законодавчий акт «Orphan Drug Act», завдяки якому вперше прозвучав термін «орфанні хвороби» та визначено 1600 рідкісних хвороб невідомої етіології. Орфанними (від англійського «orphan» – сирота, сирітський) називають захворювання, частота поширення яких не перевищує 1 випадок на 2 тис населення. Сьогодні Європейським комітетом експертів із рідкісних захворювань зареєстровано приблизно від 5 до 8 тис різних рідкісних захворювань, з них лише 250 ОЗ мають свій шифр у Міжнародній класифікації хвороб 10-го перегляду (МКХ‑10).
Вважається, що показник поширеності ОЗ не є уніфікованим. У різних країнах/регіонах критерії «орфанності» захворювання відрізняються і їх частота може коливатись. Рівень поширеності даних патологій може бути низьким в одному регіоні (чи популяції) та високим в іншому. Згідно з даними офіційного веб-сайту Європейського союзу (ЄС), в Європі будь-яке захворювання, що вражає <5 осіб на 10 тис, вважається рідкісним [1]. У більшості ж випадків захворювання вважають орфанними, коли вражається 1 людина на ≥100 тис осіб. У країнах ЄС на рідкісні захворювання страждає 6-8% населення, тобто від 27 до 36 млн людей [1]. Розрахункова поширеність орфанних ендокринних захворювань коливається від 0,08 до 64 випадків на 100 тис осіб [2] (табл. 1).

Вік пацієнтів, в якому маніфестує ОЗ, може бути різним, більшість з ОЗ діагностують у ранньому віці. Ураховуючи складнощі в діагностиці, встановлення правильного діагнозу може бути відтерміновано.
Майже 80% рідкісних патологій є результатом генних і хромосомних мутацій, інші спричинені інфекційними (віруси, бактерії), алергічними агентами або факторами довкілля. Для багатьох ОЗ характерна поліорганна ураженість, яка потребує кваліфікованої допомоги лікарів різних спеціальностей.
Таким чином, труднощі в діагностиці, дороговартісне, специфічне й часто пожиттєве лікування, неблагоприємний прогноз, висока частота інвалідизації пацієнтів зумовлюють медико-соціальне значення ОЗ.
У країнах ЄС діє спеціальна програма з вирішення різноманітних питань щодо діагностики, лікування та соціального забезпечення хворих на ОЗ. 1997 року Французьким національним інститутом охорони здоров’я та медичних досліджень створено проект «Орфанет» (www.orpha.net). На сьогодні це консорціум із 40 країн, який об’єднує інформацію стосовно ОЗ практично в усіх країнах Європи. За даними звіту Orphanet про реєстри захворювань, який опубліковано в травні 2019 року, їх загальна кількість становила: 60 європейських, 80 регіональних, 58 інтернаціональних та 535 національних реєстрів. Один із них український – це реєстр пацієнтів зі спинальною м’язовою атрофією [2].
У 2014 р. прийнято Закон України «Про внесення змін до Основ законодавства України про охорону здоров’я щодо забезпечення профілактики та лікування рідкісних (орфанних) захворювань», яким визначено поняття «рідкісне (орфанне) захворювання» – це захворювання, яке загрожує життю людини або яке хронічно прогресує, призводить до скорочення тривалості життя громадянина або до його інвалідності, поширеність якого серед населення не частіша ніж 1:2000».
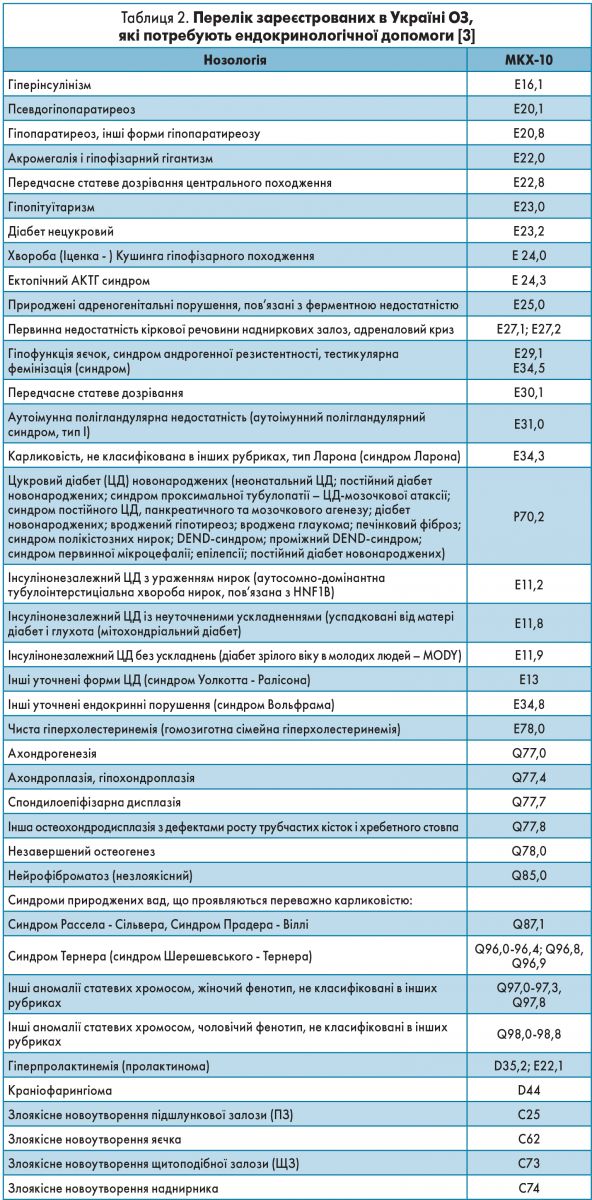 У жовтні 2014 року в Україні опубліковано наказ МОЗ № 778 «Про затвердження переліку рідкісних (орфанних) захворювань» (зі змінами, внесеними згідно з наказами Міністерства охорони здоров’я від 30.12.2015 № 919, від 29.06.2017 № 731, від 24.12.2019 № 2664). У переліку вказані ОЗ, які призводять до скорочення тривалості життя хворих або їх інвалідизації і для лікування яких є визнані терапевтичні методи. Отже, в Україні офіційно затверджено 302 нозології, віднесені до рідкісних захворювань [3]. Серед них: 61 рідкісна ендокринна хвороба (у тому числі розлади харчування та порушення обміну речовин), а також природжені вади розвитку, хромосомні аномалії та рідкісні новоутворення, у визначенні тактики ведення яких ендокринолог бере безпосередню участь [4] (табл. 2).
У жовтні 2014 року в Україні опубліковано наказ МОЗ № 778 «Про затвердження переліку рідкісних (орфанних) захворювань» (зі змінами, внесеними згідно з наказами Міністерства охорони здоров’я від 30.12.2015 № 919, від 29.06.2017 № 731, від 24.12.2019 № 2664). У переліку вказані ОЗ, які призводять до скорочення тривалості життя хворих або їх інвалідизації і для лікування яких є визнані терапевтичні методи. Отже, в Україні офіційно затверджено 302 нозології, віднесені до рідкісних захворювань [3]. Серед них: 61 рідкісна ендокринна хвороба (у тому числі розлади харчування та порушення обміну речовин), а також природжені вади розвитку, хромосомні аномалії та рідкісні новоутворення, у визначенні тактики ведення яких ендокринолог бере безпосередню участь [4] (табл. 2).
У своїй практиці лікар-ендокринолог доволі часто стикається з деякими ОЗ. Наприклад, частота виявлення пролактиноми складає 1/1600 осіб (у Бельгії, Швейцарії та інших країнах ЄС, а також у Великій Британії). На це захворювання страждають переважно жінки, особливо пременопаузального віку. Пролактинома складає 66% клінічно значущих випадків аденоми гіпофіза [5]. Лабораторні та інструментальні методи діагностики дають можливість встановити діагноз пролактиноми, провести диференційну діагностику, а наявність препаратів агоністів дофаміну – призначити коректне лікування.
У разі підозри інших ОЗ ситуація може бути іншою. Наприклад, діагностувати акромегалію (АМ) можна на момент появи клінічних проявів. Лабораторні методики визначення інсуліноподібного фактору росту – 1 (ІФР‑1), соматотропного гормону (СТГ) допомагають діагностувати гіперсоматотропінемію, а магнітно-резонансна томографія (МРТ) гіпофіза – виявити соматотропіному [6]. Сьогодні в Україні офіційно зареєстровано 844 пацієнти з АМ, хоча розрахункова загальна кількість таких пацієнтів в Україні може сягати приблизно 2500 осіб. Точнішу цифру поширеності АМ спрогнозувати складно, крім того, невідомо, скільки всього хворих на неконтрольовану АМ. Ситуація з лікуванням АМ в Україні ще складніша, чим зі статистикою, оскільки препарати для корекції гіперсоматотропінемії дороговартісні та потребують пожиттєвого призначення. Багато пацієнтів зі встановленим діагнозом не забезпечені медикаментозними препаратами, що призводить до подальшого прогресування захворювання. І це при тому, що у світову клінічну практику впроваджено цілу низку фармакологічних засобів (аналоги соматостатину, антагоністів рецепторів гормону росту та дофамінових рецепторів), які дають можливість значно підвищити ефективність комбінованого лікування аденом гіпофіза. За останні десятиліття в багатьох країнах світу були створені або створюються національні реєстри пацієнтів із різними соціально значущими захворюваннями, зокрема з АМ, що дає можливість планувати та забезпечувати хворих необхідними препаратами. Опубліковані дані бельгійських (Bex М. et al., 2007), британських (Jenkins et al., 2006), фінських (Kauppinen-Makelin et al., 2005), іспанських (Mestron et al., 2004) та інших національних реєстрів хворих на АМ. Низка інших європейських країн ініціювали створення таких реєстрів. На жаль, в Україні дотепер немає реєстру хворих на АМ, що призводить до зниження якості життя та розвитку ускладнень.
Наводимо клінічний випадок діагностики й тактики ведення хворої на АМ.
Клінічний випадок
Пацієнтка К., 1962 р.н., звернулася по консультацію в червні 2020 року зі скаргами на періодичний головний біль, виражену слабкість, підвищену втомлюваність, незначну (2 кг за 6 міс) втрату ваги.
Вважає себе хворою з 2016 року, коли почала зазначати зміну рис обличчя (збільшення хрящів носа та вушних раковин), збільшення розмірів кистей та стоп (за рік змінила кілька розмірів взуття), періодичний головний біль. З такими скаргами в травні 2017 року жінка звернулася до ендокринолога, який порекомендував лабораторне обстеження та проведення МРТ гіпофіза. За результатами лабораторного обстеження виявлено гіперсоматотропінемію (СТГ – 16,2 нг/мл, ІФР‑1 – 628 (норма 36-200) нг/мл, пролактин – норма). У процесі прицільної МРТ гіпофіза виявлено: розташування «турецького сідла» звичайне, його порожнина асиметрично розширена. Краї його дна та стінок чітко відмежовані від оточуючих тканин. Гіпофіз збільшений, розміром 15,5х19,0х9,6 мм. Диференціація долей збережена. Нейрогіпофіз без видимих змін. Структура аденогіпофіза на нативних зображеннях неоднорідна внаслідок наявності в правих відділах патологічного об’ємного утворення загальними умовними розмірами 12,0×12,7×8,6 мм, що має дещо неоднорідний ізоінтенсивний МР-сигнал на Т2WI та помірно гіпоінтенсивний МР-сигнал на Т1WI. При проведенні динамічного дослідження з внутрішньовенним болюсним введенням контрастної речовини вказане утворення з ознаками уповільненого та значно зниженого накопичення контрасту, яке зберігається на відтермінованих зображеннях, що характерно для аденоми. Латеральний контур аденоми досягає медіального контуру інтракавернозного сегмента ВСА праворуч, але ознак компресії артерії та інвазії процесу в кавернозний синус не виявлено, медіальний контур аденоми пролабує в супраселлярну цистерну, стебло гіпофіза деформоване, з девіацією ліворуч. Без відхилень від норми визначаються області перехресту зорових нервів та надсідловидної цистерни підоболонкового простору, заповненого спинномозковою рідиною. Кавернозний синус, кавернозний відділ ВСА – без видимих змін. Прилеглі відділи головного мозку не змінені.
Проведено додаткові діагностичні дослідження.
Аналіз крові: тиреотропний гормон (ТТГ) – 1,34 мОд/л (норма 0,4-4,0), тироксин вільний (Т4 вільн.) – 1,42 нг/дл (норма 0,89-1,76), пролактин – 16,5 нг/мл (норма 2,8-29,2).
Загальний аналіз крові: еритроцити – 4,44х1012/л, гемоглобін – 139 г/л, гематокрит – 37,1, тромбоцити – 215×109/л, лейкоцити – 3,2×109/л, [швидкість осідання еритроцитів] ШОЕ – 10 мм/год.
Біохімічний аналіз крові: загальний білок – 69,3 г/л (норма 65-86), сечовина – 3,4 ммоль/л (норма 2,1-8,2), креатинін – 112 мкмоль/л (норма 71-115), холестерин загальний – 4,61 ммоль/л, білірубін загальний – 20,0 мкмоль/л (норма 0-21), аланінамінотрансфераза – 12,8 Од/л (норма 0-45), аспартатамінотрансфераза – 14,9 Од/л (норма 0-40), глюкоза – 4,8 ммоль/л.
Електрокардіограма: синусова аритмія зі схильністю до сповільнення частоти серцевих скорочень (ЧСС). Електрична вісь серця відхилена вліво. Порушення провідності по правій ніжці пучка Гіса. Помірні зміни міокарда. Не виключена гіпертрофія лівого шлуночка.
Рентгенографія органів грудної клітки: інфільтративно-вогнищевої патології не виявлено.
Дослідження функції зовнішнього дихання: нормальна спірометрія.
Ультразвукове дослідження (УЗД) щитоподібної залози: щитоподібна залоза розташована в типовому місці, збільшена за рахунок обох долей. В обох долях визначаються утворення: у правій – множинні, розмірами від 4 до 11 мм, у лівій – 4, розмірами від 5 до 12 мм, правильної форми, з чіткими межами. Тканина утворень ізоехогенна. Ехоструктура неоднорідна за рахунок ділянок кістоподібної дегенерації. Інша тканина залози помірно гіпоехогенна. Ехоструктура неоднорідна через наявність множинних дрібних анехогенних включень. Регіонарні лімфатичні вузли не візуалізуються. Сумарний об’єм за методом Brunn (см3): 18,89. Права доля – 9,61 см3, ліва доля – 9,28 см3. Висновок: ехографічна картина змішаного зоба ІІ ступеня.
УЗД органів черевної порожнини. Висновок: УЗ-ознаки хронічного холециститу, хронічного панкреатиту.
На основі результатів лабораторних та інструментальних досліджень встановлено клінічний діагноз: ендосупраселлярна СТГ-продукуюча аденома гіпофіза. Синдром акромегалії. Змішаний зоб ІІ ст. Еутиреоз.
Зважаючи на наявність об’ємного утворення, з найбільшою вірогідністю аденоми гіпофіза (соматотропіноми), та враховуючи результати обстеження, пацієнтку направлено на консультацію до нейрохірурга для хірургічного лікування. 8.08.17 р. проведено трансназальне транссфеноїдальне ендокапсулярне видалення аденоми гіпофіза. Патогістологічний висновок: морфологічна картина відповідає ацидофільній аденомі гіпофіза. Післяопераційний період – без особливостей. Медикаментозне лікування не призначено.
Через 3 міс (листопад 2017 року) після оперативного втручання пацієнтка пройшла контрольне обстеження.
Результати лабораторних досліджень. СТГ – 0,4 нг/мл, ІФР‑1 – 204 нг/мл (норма 36-200), ТТГ – 0,9 мОд/л (норма 0,4-4,0), Т4 вільн. – 0,8 нг/дл (норма 0,89-1,76), пролактин – 5,4 нг/мл (норма 2,8-29,2), кортизол – 8,6 мкг/дл (норма 4,3-22,4). МРТ гіпофіза: «турецьке сідло» сплощене, розміри в межах норми. Патологічних об’ємних утворень у гіпофізарній зоні не виявлено. Гіпофіз розташований на дні «турецького сідла», злегка деформований. Розміри гіпофіза 3,8×12,0×10 мм. Воронка трохи відхилена вліво. Надселлярна група цистерн без особливостей. Хіазма не змінена. Параселлярні структури без особливостей. Висновок: стан після трансназальної транссфеноїдальної аденомектомії ендосупраселлярної аденоми гіпофіза. Ураховуючи виявлення у хворої вторинного гіпотиреозу, призначено L-тироксин 50 мкг/д.
Восени 2019 року пацієнтка почала спостерігати повернення симптомів – головний біль, збільшення розмірів стоп, виражену слабкість, підвищену втомлюваність. Приймає 50 мкг/д L-тироксину. При лабораторному обстеженні виявлено: СТГ – 3,3 нг/мл, ІФР‑1-288,4 нг/мл (норма 36-200), ТТГ – 0,6 мОД/л (норма 0,4-4,0), Т4 вільн. – 1,2 нг/дл (норма 0,89-1,76), пролактин – 6,2 нг/мл (норма 2,8-29,2), кортизол – 5,1 мкг/дл (норма 4,3-22,4). При проведенні МРТ головного мозку виявлені ознаки рецидиву аденоми гіпофіза: у селлярній ділянці візуалізується утворення неправильної форми 7×4×6 мм. У плановому порядку в січні 2020 року пацієнтці проведено повторне трансназальне транссфеноїдальне видалення аденоми гіпофіза. Операція та післяопераційний період – без особливостей. Пацієнтка продовжувала приймати L-тироксин 50 мкг/д.
Через епідеміологічну ситуацію контрольне обстеження пацієнтка пройшла в червні того ж року та звернулася за консультацією в ДУ «Інститут ендокринології та обміну речовин ім. В.П. Комісаренка НАМН України».
Об’єктивний статус. Ріст – 167 см, вага – 84 кг, [індекс маси тіла] ІМТ – 30,1 кг/м2. Тілобудова нормостенічна. Підвищеного харчування. Шкіра помірної вологості. Язик вологий, збільшений в розмірах, з відбитками зубів. Кістково-суглобовий апарат: збільшені лобні горби, вилицеві дуги, прогнатизм, діастема на верхній та нижній щелепах, збільшені ступні та кисті. Щитоподібна залоза ІІ ст., неоднорідна. Дихання везикулярне. Тони серця звучні. ЧСС – 88 уд/хв. Артеріальний тиск – 130/80 мм рт. ст. Живіт м’який, безболісний. Екзофтальму нема. Очні симптоми негативні.
Результати лабораторних досліджень. Аналіз крові: СТГ – 5,09 нг/мл, ІФР‑1 – 465,7 нг/мл (норма 36-200), ТТГ – 0,4 мОд/л (норма 0,4-4,0), Т4 вільн. – 0,98 нг/дл (норма 0,89-1,76), пролактин – 5,1 нг/мл (норма 2,8-29,2), кортизол – 6,0 мкг/дл (норма 4,3-22,4), [глікозильований гемоглобін] HbA1c – 5,4%, глюкоза – 4,93 ммоль/л. Аналіз добової сечі на кортизол – 64 мкг/добу (норма 58-403).
Консультація невропатолога. Дисциркуляторна енцефалопатія ІІ ст., астено-невротичний, цефалгічний, вестибуло-атаксичний синдроми. Поширений остеохондроз хребта.
Консультація окуліста. Ангіопатія сітківки обох очей.
Консультація гінеколога. Менопауза.
Консультація кардіолога. Ішемічна хвороба серця: стабільна стенокардія напруги 2 [функціонального класу] ФК. Атеросклеротичний кардіосклероз, атеросклероз аорти. Гіпертонічна хвороба 2 стадія, 2 ступінь, ризик 3 (високий). [серцева недостатність] СН І.
Діагноз. Рецидив аденоми гіпофіза (соматотропіноми). Акромегалія, пухлинна стадія, активна форма. Стан після повторних операцій з приводу аденоми гіпофіза (2017 і 2020 роки). Післяопераційний гіпопітуїтаризм: вторинний гіпотиреоз у стані медикаментозної компенсації. Змішаний зоб ІІ ступеня. Ішемічна хвороба серця: стабільна стенокардія напруги 2 ФК. Атеросклеротичний кардіосклероз, атеросклероз аорти. Гіпертонічна хвороба 2 стадія, 2 ступінь, ризик 3 (високий). СН І. Дисциркуляторна енцефалопатія ІІ стадії, астено-невротичний, цефалгічний, вестибуло-атаксичний синдроми. Поширений остеохондроз хребта. Хронічний холецистит у стадії ремісії. Хронічний панкреатит у стадії ремісії.
З огляду на прогресування хвороби, збереження гіперсоматотропінемії, пацієнтці рекомендовано лікування аналогами соматостатину. Проведено 3-денний тест з аналогом соматостатину короткої дії (октреотид 1,0 підшкірно 3 р./добу) з подальшим контрольним визначенням рівнів СТГ та ІФР‑1. Ураховуючи позитивний результат 3-денного тесту з октреотидом (зниження рівня ІФР‑1 з 465,7 до 267,0 15.06. і 25.06.2020 р. відповідно), пацієнтці рекомендована тривала супресивна терапія пролонгованим аналогом соматостатину (ланреотид) – 120 мг підшкірно 1 раз на 28 днів із контролем СТГ та ІФР‑1. Також рекомендовано продовжити прийом тироксину в дозі 50 мкг/д за 30 хв до сніданку.
Не менш складною буває ситуація при орфанних спадкових синдромах поєднаного пухлинного ураження різних ендокринних залоз – множинних ендокринних неоплазіях (МЕН, поліендокринопатії).
Множинні ендокринні неоплазії
Виділяють такі форми цих синдромів: МЕН1, МЕН2А (4 варіанти), МЕН2В, МЕН4, хвороба фон Гіппеля – Ліндау, нейрофіброматоз 1 типу, комплекс Карнея та синдром МакК’юна – Олбрайта.
Синдром МЕН 1 типу
МЕН тип 1 (МЕН1) – ендокринний пухлинний синдром, спричинений інактивуючими мутаціями пухлинного гена-супресора МЕN1 у 11q13 локусі. Успадковується аутосомно-домінантно, але також може виникати спорадично (без сімейного анамнезу) як результат нових мутацій. МЕН1 характеризується комбінацією паратиреоїдних пухлин, пухлин острівцевих панкреатичних клітин і пухлин аденогіпофіза. Більшість МЕН1 пухлин – неагресивні, і багато з них мають тривалий повільно прогресуючий розвиток, роками залишаючися безсимптомними. Але пацієнти з нелікованим МЕН1 мають зменшену тривалість життя з 50% вірогідністю смерті до 50 років [7].
Первинний гіперпаратиреоз (ГПТ), унаслідок гіперплазії і/або аденоми прищитоподібних залоз – найчастіший прояв МЕН1 і виникає у приблизно 90% пацієнтів [8]. Первинний ГПТ при МЕН1 має тривалий безсимптомний перебіг і зазвичай діагностується як випадкова знахідка підвищеного рівня паратгормона в плазмі в пацієнтів із гіперкальціємією і, у деяких випадках, з нормокальціємією. Клінічні прояви можуть включати гіперкальціємію, нефролітіаз, порушення будови кісткової тканини (генералізована фіброзна остеодистрофія).
Первинний ГПТ у складі МЕН1 відрізняється від не-МЕН1 первинного ГПТ наступним [9]:
- Молодший вік початку захворювання (20-25 років).
- Залученість декількох прищитоподібних залоз.
- Високий рівень рецидиву гіперкальціємії після паратиреоїдектомії (50% через 8-12 років після оперативного втручання).
Пухлини острівцевих клітин ПЗ представляють другий найбільш частий прояв МЕН1, виникаючи в 30-80% пацієнтів, та можуть бути представлені наступними варіантами [7]:
- Гастриноми.
- Інсуліноми.
- Глюкагономи.
- Вазоактивні інтестинальні поліпептидоми (ВІПоми).
- Панкретичні поліпептидоми (ППоми).
Для цих пухлин характерна мультицентричність і здатність до злоякісного переродження. Найчастіше пухлини ПЗ у складі МЕН1 є гормонально неактивними. Вони або не виділяють гормони, або виділяють гормонально неактивні пептиди, такі як панкреатична поліпептидаза, хромогранін А, нейротензин, нейро-специфічна енолаза, або грелін. Гормонально неактивні пухлини ПЗ, особливо малого розміру (<2 см), важко діагностувати стандартними візуалізаційними методами. У таких випадках ефективним є чутливий радіологічний скринінг. Вчасна діагностика цих пухлин дуже важлива, тому що в пацієнтів до 80 років пенетрантність панкреатичних пухлин складає >80%, а найчастішою причиною смертності, пов’язаної з МЕН1, є метастатичне ураження [10, 11].
Гастриноми – найбільш часті серед гормонально активних панкреатичних нейроендокринних пухлин і виникають у 40-55% пацієнтів [12]. Розвитку гастрином передує багатофокусна гіперплазія гастрин-продукуючих клітин. Якщо порівняти з не-МЕН1 гастриномами при синдромі Золінгера – Еллісона (ЗЕС), гастриноми при МЕН1 часто невеликого розміру (<0,5 см), мультицентричні та локалізовані в дванадцятипалій кишці, зменшуючи вірогідність успішного хірургічного лікування. Приблизно 20-30% пацієнтів із ЗЕС мають МЕН1. У хворих на МЕН1 ЗЕС з’являється тільки в осіб із первинним ГПТ [7]. Тривалий МЕН1 і ЗЕС можуть призвести до розвитку карциноїдних пухлин шлунка, у деяких випадках – високоагресивних [13].
Інсуліноми – на другомі місці за частотою з гормонально активних панкреатичних нейроендокринних пухлин при МЕН1. Вони розвиваються в молодому віці (<35 років) у приблизно 10-30% пацієнтів. Інсуліноми при МЕН1 можуть маніфестувати як одиничні макроаденоми ПЗ (>2 cм) або, частіше, як множинні мікроаденоми (<2 см), розкидані по всій ПЗ [14]. Інсуліноми бувають мультицентричними і можуть метастазувати в регіонарні лімфовузли або печінку [7].
Глюкагономи в пацієнтів із МЕН1 виникають рідко (<3%) і можуть протікати без змін вуглеводного обміну або з гіперглікемією. У деяких пацієнтів спостерігається типове ураження шкіри, відоме як некролітична мігруюча еритема. Іншими симптомами можуть бути: анемія, стоматит, втрата ваги, але часто вони відсутні.
У менше ніж 1% пацієнтів із МЕН1 у ПЗ виникають ВІПоми, пухлини, які секретують панкреатичний поліпептид, та пухлини, що виділяють гормон росту [7].
МЕН1-асоційовані пухлини аденогіпофіза найчастіше секретують пролактин (60%) і гормон росту (25%). Менше ніж 5% – кортикотропін, інші пухлини – гормонально неактивні [7]. Проти не-МЕН1 пухлин аденогіпофіза МЕН1 пухлини аденогіпофіза є більшими (макроаденоми) і агресивнішими, з більшим рівнем інфільтрації пухлинних клітин у нормальну тканину аденогіпофіза. МЕН1-асоційовані пухлини аденогіпофіза важче піддаються лікуванню. При цьому вираженої гістологічної різниці між МЕН1 і не-МЕН1 пухлинами аденогіпофіза немає [15].
Також у пацієнтів із МЕН1 можуть виникати карциноїдні пухлини в бронхах, шлунково-кишковому тракті, ПЗ та тимусі. Карциноїд тимуса, асоційований з МЕН1, часто гормонально неактивний і високоагресивний [16]. Карциноїди можуть активно секретувати серотонін, соматостатин, кортикотропін і гормон росту.
Для МЕН1 характерні шкірні прояви. Підшкірні ліпоми виявляються в третини пацієнтів із МЕН1. Ці новоутворення мають втрату гетерозиготності в сегменті 11q12-12 і асоційовані з дефективним функціонуванням глобулярного протеїну. Ліпоми при МЕН1 можуть бути ретроперитонеальними, вісцеральними або плевральними. Наявність ангіофібром і колагеном на обличчі дає можливість досимптомного встановлення діагнозу МЕН1 у родичів пацієнта з встановленим діагнозом [7].
Пухлини надниркових залоз виникають у 20-40% пацієнтів із МЕН1. Зазвичай ці пухлини доброякісні і представлені гормонально неактивними кортикальними аденомами чи дифузною або нодулярною гіперплазією [17, 18]. Адренокортикальні карциноми при МЕН1 зазначаються рідко.
Аденома ЩЗ розвивається в 5-30% пацієнтів і не має специфічних, пов’язаних із МЕН1, ознак.
Спостерігаються менінгіоми та інші пухлини центральної нервової системи (ЦНС).
Нейроендокринна пухлина тимуса – рідкісний (3,7% пацієнтів із МЕН1), але фатальний компонент МЕН1 – на його рахунку майже 20% МЕН1-асоційованої смертності [19].
Ген, який відповідає за розвиток МЕН1, локалізований у сегменті 11q13 і кодує білок менін, який задіяний у регуляції транскрипції і геномної стабільності. Втрата гетерозиготності цієї ділянки асоційована з МЕН1, що дає підстави вважати, що ген має пухлино-супресорну функцію. Пацієнти успадковують одну мутовану копію гена, і їм залишається отримати соматичну мутацію другої копії для розвитку пухлини. МЕН1 – аутосомно-домінантне захворювання, але спорадичні мутації виникають також. Ідентифікація та генетична характеристика причинного гена відкрили можливість генетичного тестування і ранньої діагностики захворювання. Аналіз послідовності в гені МЕN1 для виявлення мутацій забезпечує найточніше підтвердження статусу носія гена. З розвитком нових технологій, таких як мультиплексна ампліфікація лігованих зондів (MLPA), були відкриті нові мутації в гені МЕN1, що підвищує чутливість генетичного аналізу. У минулому генетичне тестування не знаходило мутації МЕN1 у 10-30% пацієнтів із клінічними критеріями для діагнозу МЕН1 (наявність щонайменше двох МЕН1-асоційованих пухлин: первинний ГПТ, дуоденопанкреатичні нейроендокринні пухлини, пухлини аденогіпофіза) [20]. Таких пацієнтів називають фенокопіями. Наявність фенокопії передбачається при негативному тесті МЕN1 за послідовністю, генною дозою і аналізом гаплотипу 11q13. Фенокопії нараховують до 5% МЕН1-подібних випадків, більшою мірою пов’язаних із захворюваннями прищитоподібних залоз і аденогіпофіза [21].
Згідно з результатами рандомізованих досліджень частота МЕН1 складає від 1 випадку на 10 тис до 1 випадку на 100 тис осіб [7, 22]. Характерне формування географічних кластерів як наслідок ефекту засновників (у популяційній генетиці – явище втрати генетичної мінливості при формуванні нової популяції невеликою кількістю людей з більшого числа населення) [23]. Ген МЕН1 має високу пенетрантність. У віковій когорті пацієнтів до 20 років ген пенетрантний у 50% осіб, до 40 років – у 95%. Не було ідентифіковано випадків типових ознак, асоційованих зі статусом носія гена МЕН1 у пацієнтів молодше 5 років, таким чином, ген не пенетрантний до цього віку [7, 8].
Тактика лікування залежить від наявних ендокринопатій та стратифікації ризиків. Основним методом лікування пацієнтів із МЕН1 є хірургічне видалення пухлин, у низці випадків показані превентивні втручання [24].
У нелікованих пацієнтів із МЕН1 знижена тривалість життя з 50% вірогідністю смерті у віці до 50 років. Причини смерті пов’язані зі злоякісними пухлинами або наслідками ендокринних порушень [7]. Злоякісні нейроендокринні пухлини ПЗ або карциноїдні пухлини тимуса пов’язані зі зростанням ризику смерті в пацієнтів із МЕН1. Приблизно 70% пацієнтів із МЕН1 помирають від причин, прямо пов’язаних із МЕН1 [12].
Синдром МЕН 2 типу
МЕН тип 2 (МЕН2) – спадкове захворювання, яке характеризується розвитком медулярної карциноми ЩЗ, пухлин прищитоподібних залоз і феохромоцитоми. МЕН2 виникає внаслідок мутацій у RET-протоонкогені і передається аутосомно-домінантно.
Є два МЕН2 синдроми: МЕН2А і МЕН2В (раніше мав назву МЕН3).
МЕН2А далі класифікується на 4 варіанти, відповідно до наявності його складових. Класичний МЕН2А характеризується медулярною карциномою ЩЗ, первинним ГПТ і феохромоцитомою. Три додаткові варіанти складають: МЕН2А з вузликовим амілоїдозом шкіри, МЕН2А з хворобою Гіршпрунга і сімейна медулярна карцинома, яка діагностується, коли в пацієнта є патологічний RET геном і медулярна карцинома, але в сімейному анамнезі відсутні феохромоцитома або гіперпаратиреоз [25].
МЕН2В зазначається не так часто, як МЕН2А, нараховуючи 5% випадків МЕН2. Він характеризується більш агресивною медулярною карциномою (яка виникає практично в 100% випадків), феохромоцитомою (50%), мукозними невромами (95%-98%) і гангліоневромами (40%). Гіперпаратиреоз відсутній. Крім того, майже в усіх пацієнтів виявляється марфаноїдна тілобудова: високе піднебіння, лійковидна деформація грудної клітки, високе склепіння стоп, сколіоз, виступаючі губи. Часто виникають невроми на повіках, кон’юнктиві, слизовій носа та гортані, язику та губах [26].
Медулярна карцинома розвивається фактично в усіх пацієнтів із МЕН2. Найчастіше це перший прояв синдрому, який виникає впродовж 1-3-ї декади життя. У пацієнтів із МЕН2А медулярна карцинома зазвичай двостороння і багатофокусна, її розвитку передує гіперплазія С-клітин, на відміну від спорадичної медулярної карциноми, яка є односторонньою [27].
Обґрунтування генотип-фенотипової кореляції спадкової медулярної карциноми може скерувати розвиток індивідуального підходу до оцінки й контролю ризиків у дітей – носіїв генотипу. Для довгострокового запобігання розвитку медулярної карциноми в пацієнтів із МЕН2 є ефективною рання тотальна тиреоїдектомія [28-30].
Феохромоцитоми присутні майже в 50% МЕН2А пацієнтів. Вони двосторонні в 60-80% випадків, у той час як спорадичні феохромоцитоми двосторонні в 10%. Феохромоцитоми діагностуються одночасно з медулярною карциномою або через декілька років. Феохромоцитоми у складі МЕН2А майже завжди доброякісні. Але вони можуть спричиняти загрозливі для життя гіпертонічні кризи або напади аритмії.
Гіперпаратиреоз присутній приблизно в половини пацієнтів із МЕН2А, але виявляється рідше за феохромоцитоми. Найчастіше проявляється в пацієнтів після 30 років. Гістологічно прищитоподібні залози при МЕН2А складаються з гіперплазованих головних клітин, ця гіперплазія є асиметричною відносно розмірів прищитоподібної залози. Спостерігається підвищена частота додаткових прищитоподібних залоз [31]. Персистуючий або рецидивуючий гіперпаратиреоз не характерний для МЕН2А, на відміну від пацієнтів із МЕН1.
МЕН2 спричиняють мутації в трансмембранному протоонкогені RET, локалізовані в 10q11.21. Функція білка, закодованого RET, залишається невизначеною, але відомо, що він є важливим під час ембріонального розвитку нервової системи кишечнику й нирок. RET складається з трьох доменів: домену цистеїнвмісного позаклітинного рецептора, гідрофобного трансмембранного домену і внутрішньоклітинного тирозинкіназного каталітичного домену [32, 33].
Позаклітинний домен взаємодіє з одним із відомих сьогодні чотирьох лігандів. Ці ліганди – гліальна клітинна лінія – похідне нейротропного фактору (GDNF), нейротурин, персефін і артемін – також взаємодіють з одним із чотирьох корецепторів сімейства рецепторів – GDNF-альфа-сімейством. GDNF відіграє важливу роль у нормальному функціонуванні шляхів, задіяних у нейрогенезі нервової системи кишечнику і нирковому органогенезі. Тирозинкіназний каталітичний центр знаходиться у внутрішньоклітинному домені і викликає низхідні сигнальні впливи через різні молекули – вторинні месенджери [34].
Точкові мутації, асоційовані з МЕН2А і її підтипом – сімейною медулярною карциномою, ідентифіковані в екзонах 10 і 11. Доведена генотипно-фенотипна кореляція. Класична МЕН2А асоційована з міссенс-мутацією зародкової лінії в кодонах 609, 611, 618 або 620 екзону 10 або кодону 634 екзону 11 у RET-протоонкогені. МЕН2А з вузликовим амілоїдозом шкіри майже завжди асоційована з мутацією кодону 634, тоді як пацієнти з МЕН2А і хворобою Гіршпрунга типово мають мутації в екзоні 10 RET [35].
Є дані, що в пацієнтів із мутацією в кодоні А883F більш латентний розвиток захворювання проти носіїв M918T, з більш пізньою маніфестацією феохромоцитом [36].
Близько 75% випадків МЕН2В спорадичні, і пацієнти мають RET-мутації de novo, тоді як 25% випадків виникають у родинах із проявами МЕН2В у минулому або зараз. Майже 95% пацієнтів із МЕН2В мають мутацію зародкової лінії RET в екзоні 16 (кодон M918T), і менше ніж 5% – в екзоні 15 (кодон A883F) [25].
Загальна частота МЕН2 складає 1 випадок на 30-50 тис осіб [37, 38].
Найчастішими видами МЕН2 є MEН2A, MEН2A із сімейною медулярною карциномою і МЕН2B.
У пацієнтів молодших за 50 років із МЕН2А з мутацією гена RET захворювання виявляється в 50%, у віці до 70 років – у 70%. Медулярна карцинома в пацієнтів із МЕН2В спостерігається в ранньому дитячому віці.
Лікування МЕН2 – хірургічне. До проведення паратиреоїдектомії або тиреоїдектомії обов’язково потрібно виключити феохромоцитому. Якщо феохромоцитома діагностована, втручання з її приводу проводиться першочергово, з огляду на можливу гемодинамічну нестабільність [39, 40]. При метастатичних ураженнях можуть бути застосовані інгібітори тирозинкінази.
При МЕН2 раннє лікування медулярної карциноми ЩЗ може запобігти смерті, а ретельний моніторинг феохромоцитоми – зменшити ризик гіпертензивних кризів. Ураховуючи, що пенетрантність медулярної карциноми при МЕН2 складає майже 100%, у пацієнтів із високим ризиком мутацій RET профілактична тиреоїдектомія показана в немовлячому віці, а в дітей з виявленими мутаціями RET – у віці до 5 років [41, 42].
Синдром МЕН 4 типу
МЕН тип 4 (МЕН4) має клінічні прояви, подібні до МЕН1, але викликані мутаціями в іншому гені. Найбільш типовим проявом МЕН4 є гіперпаратиреоз, на другому місці – пухлини аденогіпофіза. Ризик розвитку панкреатичних нейроендокринних пухлин у пацієнтів із МЕН4 значно нижчий за такий у пацієнтів із МЕН1 [43].
При МЕН4 виникають мутації в гені циклін-залежного інгібітора кінази (CDKN1B). Цей ген відповідає за синтез білка p27. Як і менін (який кодується геном МЕN1), р27 – пухлинний супресор, що допомагає контролювати ріст і ділення клітин. Мутації в гені CDKN1B зменшують кількість функціонуючого р27, що призводить до безконтрольного росту й розмноження клітин. Таке некероване ділення клітин може спричинити розвиток пухлин в ендокринних залозах та інших тканинах.
Задокументовано 12 підтверджених випадків МЕН4, за розрахунковими даними, її частота в загальній популяції може складати менше 1 випадку на 1 млн осіб.
Хвороба фон Гіппеля – Ліндау
Хвороба фон Гіппеля – Ліндау (ФГЛ) – рідкісне генетичне захворювання, що характеризується вісцеральними кістами і доброякісними пухлинами в багатьох органах, які мають потенціал до злоякісного переродження. Клінічні прояви хвороби ФГЛ включають розвиток гемангіом сітківки та ЦНС, феохромоцитом, множинних кіст у ПЗ та нирках і підвищений ризик трансформації ниркових кіст у рак нирки [44, 45]. Широкий діапазон віку і різноманітність форм, в яких проявляється хвороба ФГЛ, утруднює діагностику й лікування уражених пацієнтів, а також їхніх родичів. Гемангіоми сітківки діагностуються в 50% пацієнтів із хворобою ФГЛ. Пухлини ЦНС та інших органів – у 25%. Такі пацієнти мають підвищений ризик розвитку феохромоцитом, які можуть бути двосторонніми. Феохромоцитоми виникають у 10-50% пацієнтів із хворобою ФГЛ, залежно від субтипу, зумовленого видом мутації в причинному гені. Більшість (70%) панкреатичних уражень є простими кістами, які рідко бувають із клінічними проявами або перероджуються в злоякісні пухлини.
Хвороба ФГЛ успадковується аутосомно-домінантно. Нові мутації виникають у приблизно 1 немовляти з 4,4 млн новонароджених і складають 20% випадків. Загальна частота хвороби ФГЛ у світі – 1:32 000 новонароджених, у США – 1:36 000 новонароджених. Вік встановлення діагнозу коливається від раннього дитячого до 60-70 років і в середньому складає 26 років [46].
Ген фон Гіппеля – Ліндау – VHL – розміщений на короткому плечі 3-ї хромосоми (3р25.3) і кодує широко експресовану іРНК, що кодує 3 екзони з альтернативним сплайсингом [47]. У результаті 2 синтезованих білки VHL (pVHL), 30кДа форма (30р) і 19 кДа форма (р19) складають човниковий механізм між ядром та цитоплазмою, де вони утворюють комплекси з іншими білками. Ці білкові комплекси мають багато функцій: регуляція старіння, кисне-чутливий шлях, мікротубулярна стабільність і орієнтація, формування війок, цитокінова сигнальна система, збирання колагену IV типу в позаклітинному матриксі, регуляція збирання нормального позаклітинного фібронектинового матриксу і пухлинна супресія [46].
Середня тривалість життя в пацієнтів із хворобою ФГЛ складає 49 років, передусім через високу частоту асоційованого світлоклітинного раку нирки. Але ретельні обстеження, які допомагають провести ранні втручання, здатні подовжити тривалість життя таких пацієнтів.
Нейрофіброматоз 1 типу
Нейрофіброматоз 1 типу (НФ1) – мультисистемне генетичне захворювання, яке характеризується шкірними проявами у вигляді плям кольору «кави з молоком» і пахвовим лентіго, кістковими дисплазіями і розвитком доброякісних і злоякісних пухлин нервової системи, найчастіше нейрофібром. Ендокринологічний інтерес у НФ1 представляють феохромоцитоми, які спостерігаються в приблизно 5% пацієнтів [48, 49].
НФ1 виникає внаслідок мутації або делеції в гені NF1. Нейрофібромін, який кодується геном NF1, є пухлинним супресором, зменшена продукція цього білка результує численими клінічними проявами [50].
Розрахункова частота НФ1 складає 1 випадок на 3000, але насправді може бути більшою через неповне виявлення легких форм захворювання. Майже 50% пацієнтів представляють перший випадок у сім’ї як результат нової генетичної мутації [49].
Середня тривалість життя пацієнтів із НФ1 скорочується на 8 років, якщо порівняти із загальною популяцією [51].
Комплекс Карнея
Комплекс Карнея – аутосомно-домінантний синдром, який проявляється плямистою пігментацією шкіри, ендокринопатіями, ендокринними та неендокринними пухлинами [52]. Ці пухлини включають міксоми шкіри, серця, молочних залоз та інших локалізацій: первинну пігментовану нодулярну адренокортикальну хворобу (PPNAD), псаммоматозні меланотичні шваноми; аденоми аденогіпофіза, що продукують гормон росту, тестикулярні пухлини клітин Сертолі, пухлини ЩЗ, аденоми проток молочних залоз, АМ внаслідок соматомамотрофної гіперплазії та аденом, незалежних від соматоліберину.
Комплекс Карнея успадковується аутосомно-домінантно з варіативною пенетрантністю. Вважається, що кардіальні міксоми виникають із субендотендокардіальних мезенхімальних мультипотентних клітин-попередників. Комплекс Карнея виникає внаслідок мутацій у гені PRKAR1A, який кодує регуляторну субодиницю протеїнкінази А R1α [53]. PRKAR1A може грати роль гена – пухлинного супресора, регулюючи активність протеїнкінази А, що, своєю чергою, може стимулювати або пригнічувати ріст і диференціювання клітин. Гени комплексу Карнея асоційовані з геномною нестабільністю, клітинні лінії, отримані з пухлин комплексу Карнея, накопичують хромосомні перебудови, включаючи асоціації теломер і дицентричні хромосоми [54].
Пухлини PPNAD характеризуються ліпофусцин-вмісними, кортизол-продукуючими вузлами, які оточені атрофованою адренокортикальною і нормальною адреномедулярною тканиною. Природа виникнення цих пухлин незрозуміла [55]. Макроскопічно поверхня вкрита множинними маленькими вузликами чорного або коричневого кольору. Позапухлинна тканина кори надниркових залоз атрофічна. Мікроскопічно множинні вузлики визначаються в усій товщі кори. Клітини в цих вузликах позитивно фарбуються реактивом Шиффа. Поза корою зазначається інтенсивна дезорганізація без нормального зонування.
У приблизно 75% пацієнтів із комплексом Карнея спостерігається багатовузловий зоб.
З моменту виділення комплексу Карнея в окрему клінічну категорію в 1985 р. цей діагноз був встановлений приблизно 600 пацієнтам в усьому світі [56].
Середня тривалість життя хворих із комплексом Карнея становить 50-55 років. 25% смертності пацієнтів із цим синдромом зумовлені кардіальними міксомами. Тривалість їхнього життя може бути подовжена за умови регулярних обстежень і своєчасних втручань [57].
Синдром МакК’юна – Олбрайта
Синдром МакК’юна – Олбрайта діагностується за наявності щонайменше двох з наступних трьох критеріїв: поліоссальна фіброзна остеодисплазія; пігментація шкіри у вигляді плям кольору «кави з молоком»; автономна ендокринна гіперфункція (наприклад, гонадотропін-незалежне передчасне статеве дозрівання). Також можуть бути присутніми інші ендокринні синдроми, зокрема гіпертиреоз, АМ, синдром Кушинга, гіперпролактинемія, гіперпаратиреоз, гінекомастія і гіпофосфатемічна остеомаляція [58].
Синдром МакК’юна – Олбрайта розвивається внаслідок постзиготичної активуючої мутації гена GNAS1 у локусі 20q13.1-13.2, який кодує протеїн G субодиниці Gs альфа в уражених тканинах. Клінічні прояви синдрому дуже варіативні, залежно від того, які з можливих компонентів домінують [59].
Синдром МакК’юна – Олбрайта – дуже рідкісне спорадичне захворювання. Його частота в популяції оцінюється між 1 випадком на 100 тис і 1 випадком на 1 млн осіб [60].
Тяжкі випадки синдрому із залученням множинних ендокринних тканин розпізнаються в період новонародженості, як і випадки синдрому Кушинга і гіпертиреозу. Менш тяжкі форми проявляються в ранньому дитинстві від 3 міс до 9 років. Аденоми гіпофіза, що продукують гормон росту і токсичні аденоми ЩЗ внаслідок GNAS1 мутацій, можуть виникати в будь-якому віці. Захворювання з більш пізнім дебютом асоціюються з клінічно більш латентними фенотипами [61].
На сьогодні немає лікування, спрямованого на причинну молекулярну проблему синдрому. Застосовуються різні препарати для корекції різних ендокринних та метаболічних порушень, у тому числі інгібітори ароматази, стероїди, аналоги соматостатину, агоністи дофаміну, бісфосфонати, антагоністи рецепторів естрогену, антитиреоїдні медикаментозні засоби.
При переломах кісток внаслідок диспластичного ураження показане хірургічне втручання. За наявності вузлового або багатовузлового зобу з тиреотоксикозом показане проведення гемітиреоїдектомії або тиреоїдектомії. Лікування синдрому Кушинга у складі синдрому МакК’юна – Олбрайта передбачає двосторонню адреналектомію з подальшою стероїдною замісною терапією [62].
Окремо від невеликої підгрупи пацієнтів із підвищеною периопераційною летальністю і зі злоякісними новоутвореннями синдром МакК’юна – Олбрайта не пов’язаний з підвищеною смертністю. Захворюваність і втрата працездатності в таких пацієнтів може бути високою через хронічний біль і деформації кісткової тканини, а також мультигормональні ендокринопатії. Таким чином, прогноз більшою мірою залежить від проявів синдрому МакК’юна – Олбрайта [63].
Ураховуючи, що МЕН є мультисистемними захворюваннями з низкою різноманітних проявів, тактика ведення таких пацієнтів є нестандартною і потребує мультидисциплінарного підходу.
Генетичне тестування – базовий елемент у діагностиці МЕН-синдромів. Генетичний скринінг мутацій проводиться в пацієнтів із відповідними клінічними критеріями. Якщо мутація ідентифікована, тим членам сім’ї, які зазнають ризику, також проводять скринінг. Ретельні динамічні обстеження пацієнтів із МЕН, а також безсимптомних носіїв дають можливість виявляти новоутворення на ранній досимптомній стадії і своєчасно провести оперативне лікування, що може зменшити ризик пов’язаної смертності.
Останнім часом прослідковується тенденція до збільшення частоти виявлення ОЗ. Причиною може бути покращення методів лабораторної та інструментальної діагностики, використання генетичних методів діагностики та більш широка обізнаність лікарів щодо ОЗ.
У хірургічному відділі ДУ «Інститут ендокринології та обміну речовин імені В.П. Комісаренка НАМН України» з 1996 по 2020 рік пройшли лікування 29 пацієнтів із множинними ендокринними неоплазіями. МЕН1 була діагностована в 10 пацієнтів, МЕН 2А – у 13, МЕН2В – у 5 і синдром фон Гіппеля – Ліндау – в 1 пацієнта.
У травні 2020 року МОЗ України ініціювало ухвалення Національної стратегії з профілактики, діагностики та лікування рідкісних (орфанних) захворювань, що свідчить про визнання проблеми на державному рівні. Таким чином, не лише визначений перелік ОЗ, але й прийнято рішення щодо важливості лікування рідкісних (орфанних) захворювань та забезпечення пацієнтів життєво необхідними лікарськими засобами та спеціальним харчуванням. Для реалізації Національної стратегії важливим є збір статистичної інформації про ОЗ в Україні, впровадження ефективних методів ранньої діагностики, лікування та реабілітації хворих, а також підготовка кваліфікованих фахівців. Шлях вирішення цього питання – це забезпечення доступу пацієнтів до високоспеціалізованої медичної допомоги, розробка вітчизняних стандартів з імплементацією міжнародних протоколів діагностики та лікування ОЗ, проведення наукових досліджень у сфері рідкісних захворювань, а також поглиблення національної і міжнародної співпраці в питаннях тактики ведення цієї когорти пацієнтів.
Список літератури знаходиться в редакції.
Тематичний номер «Діабетологія, Тиреоїдологія, Метаболічні розлади» № 3 (51) 2020 р.
