


15 січня, 2021
Вроджені коагулопатії: сучасні погляди на діагностику та лікування
 Сьогодні до вроджених коагулопатій належать такі захворювання: гемофілія А та В, хвороба Віллебранда й рідкісні дефіцити факторів згортання крові (І, ІІ, V, VII, X, XI, XII та XIII). Серед них найпоширенішим захворюванням є гемофілія А (рис.). Термін «гемофілія» вперше застосував Friedrich Hopff у 1828 р. [1]. Сучасне тлумачення терміна «гемофілія» включає два основних захворювання: гемофілія А, коли виявляють дефіцит фактора згортання крові людини VIII (FVIII), та гемофілія В при дефіциті фактора IX (FIX). Гемофілія А – найтяжча форма гемофілії, яка становить за різними даними 80-85% від загальної кількості усіх випадків гемофілії, гемофілія В – 15-20% усіх випадків. Поширеність гемофілії А серед новонароджених оцінюють на рівні 24,6 на 100 000 хлопчиків (9,5 випадку тяжкої гемофілії А), гемофілії В – 5,0 на 100 000 хлопчиків (1,5 випадку тяжкої гемофілії В).
Сьогодні до вроджених коагулопатій належать такі захворювання: гемофілія А та В, хвороба Віллебранда й рідкісні дефіцити факторів згортання крові (І, ІІ, V, VII, X, XI, XII та XIII). Серед них найпоширенішим захворюванням є гемофілія А (рис.). Термін «гемофілія» вперше застосував Friedrich Hopff у 1828 р. [1]. Сучасне тлумачення терміна «гемофілія» включає два основних захворювання: гемофілія А, коли виявляють дефіцит фактора згортання крові людини VIII (FVIII), та гемофілія В при дефіциті фактора IX (FIX). Гемофілія А – найтяжча форма гемофілії, яка становить за різними даними 80-85% від загальної кількості усіх випадків гемофілії, гемофілія В – 15-20% усіх випадків. Поширеність гемофілії А серед новонароджених оцінюють на рівні 24,6 на 100 000 хлопчиків (9,5 випадку тяжкої гемофілії А), гемофілії В – 5,0 на 100 000 хлопчиків (1,5 випадку тяжкої гемофілії В).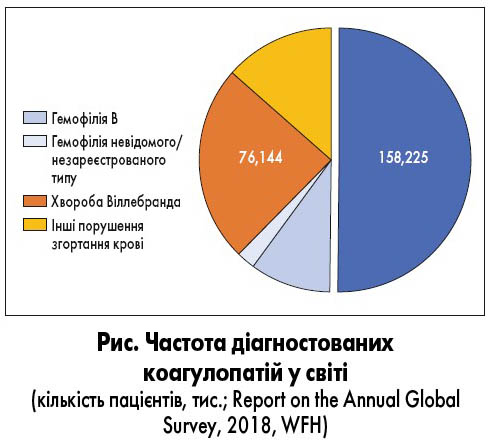
Гемофілія зазвичай успадковується через Х-хромосому з мутацією гена F8 або F9. Проте гени F8 та F9 схильні до нових мутацій, тому близько 30% усіх випадків є результатом спонтанних генетичних варіацій. За даними проспективних досліджень, понад 50% людей з уперше діагностованою гемофілією тяжкої форми не мають цього захворювання в сімейному анамнезі [2].
Залежно від мутації, яка визначає рівень і активність FVIII, хворих на гемофілію можна розподілити на три категорії з великим впливом на ризик виникнення антитіл до FVIII [3]:
- пацієнти з CRM (антиген)-негативним статусом (близько 50%), у яких втрачений антиген FVIII, що його можна виявити імунологічними методами, наприклад, у разі дефекту мРНК і нонсенс-опосередкованого розладу синтезу FVIII;
- пацієнти з CRM (антиген)-позитивним статусом (близько 5%) зі значним рівнем білка FVIII, але імовірно порушеною його функцією, викликаною місенс або іншими дрібними мутаціями;
- проміжна група пацієнтів (близько 45%) зі зниженим вмістом CRM (антигену), можливо, внаслідок неефективної секреції або прискореного розщеплення FVIII.
Важливо, що низький рівень FVIII може залежати не тільки від мутації F8 або його промотору, а й від дефектних білків, які беруть участь у його секреції. Інший фенотип гемофілії був виявлений у пацієнтів з мутацією гена vWF, що впливає на його зв’язування з FVIII, тим самим знижуючи напіврозпад останнього від нормальних 12 до 1 год. Аналіз мутацій при гемофілії А надає корисну інформацію не лише для підтвердження хвороби або визначення статусу носіїв і пренатальної діагностики, а й для прогнозування фенотипу та ризику розвитку антитіл проти екзогенного фактора VIII, а також для вибору оптимального терапевтичного режиму [4].
Гемофілію, як правило, мають лише особи чоловічої статі, які успадковують змінену материнську Х-хромосому. У жінок гемофілія (рівень FVIII або FIX <40 МО/дл) зустрічається рідко; в таких випадках зміненими є обидві Х-хромосоми; жінка з однією зміненою хромосомою вважається носієм гемофілії.
Характерним фенотиповим проявом гемофілії є схильність до кровотеч, тяжкість яких загалом корелює зі ступенем дефіциту факторів коагуляції. Особи з гемофілією легкого ступеня не обов’язково мають проблеми у вигляді аномальних або тривалих кровотеч, доки не перенесуть серйозну травму або хірургічне втручання. Найчастіше відбуваються внутрішньосуглобові та внутрішньом’язові крововиливи, а також крововиливи у внутрішні органи. У новонароджених і дітей молодших 2 років з тяжкою формою гемофілії поширеними типами кровотеч є крововиливи в м’які тканини (внутрішньом’язові), кровотечі, пов’язані з медичними процедурами (наприклад венепункція, встановлення центрального катетера, обрізання крайньої плоті, п’ятковий тест), крововиливи у шкіру та слизові оболонки (наприклад, ротової та носової порожнин), екстракраніальні крововиливи. Деякі типи кровотеч можуть бути небезпечними для життя і потребують негайної медичної допомоги та лікування.
Діагностика гемофілії ґрунтується на таких принципах [5]:
- розуміння клінічних особливостей гемофілії та відповідності клінічного діагнозу;
- використання скринінгових тестів, таких як протромбіновий час (ПЧ) і активований частковий тромбопластиновий час (АЧТЧ) або аналіз функцій тромбоцитів, для виявлення потенційних причин кровотечі (маючи на увазі, що нормальні результати скринінгових тестів не виключають можливості клінічно значущого порушення гемостазу);
- підтвердження діагнозу визначенням рівня факторів коагуляції крові та іншими необхідними специфічними дослідженнями.
Комплексне лікування при гемофілії включає багатопрофільні медичні послуги, необхідні для діагностики, лікування захворювання та його ускладнень. Такі послуги зазвичай надаються центрами лікування гемофілії або центрами комплексної медичної допомоги при гемофілії. Комплексне лікування дозволяє досягнути фізичного і соціально-психологічного здоров’я та високої якості життя хворих на гемофілію, а також знижує ризик ускладнень і смерті. Воно має охоплювати усіх членів родини, у тому числі діагностику та клінічне ведення носіїв. Пріоритетними завданнями лікування та догляду при гемофілії є профілактика крововиливів та ураження суглобів; швидке зупинення кровотечі, фізична терапія та реабілітація після гемартрозів; усунення болю; лікування ускладнень з боку опорно-рухового апарату; профілактика та лікування інгібіторної форми; лікування супутніх захворювань; догляд за зубами; оцінювання якості життя та соціально-психологічна допомога; генетичне консультування та діагностика; постійне навчання та підтримка пацієнтів і членів їх родин, які здійснюють догляд [6].
Для лікування гемофілії доступні різні типи гемостатичних засобів і препаратів для коагуляції. Широкий асортимент класів і типів продуктів, що використовуються у всьому світі, відображає еволюцію засобів для лікування гемофілії та відмінності у місцевих ресурсах і можливостях системи охорони здоров’я. Концентрати факторів згортання крові (КФК) є найкращим методом лікування хворих на гемофілію, оскільки вони безпечні й ефективні для лікування та профілактики кровотеч. Наявні два основних типи КФК: вірусінактивовані продукти, отримані з плазми крові, та рекомбінантні продукти, виготовлені з застосуванням генної інженерії та рекомбінантних технологій. Нещодавня розробка методів лікування з використанням нефакторних замісних препаратів, таких як еміцизумаб, забезпечила альтернативний підхід до лікування [7].
КФК FVIII доступні у флаконах, на яких нанесена активність продукту в міжнародних одиницях (у діапазоні від 250 до 3000 МО на флакон). Кожна 1 МО плазмового або рекомбінантного FVІІІ на 1 кг маси тіла підвищує рівень FVІІІ у плазмі крові приблизно на 2 МО/дл. Стандартний період напіввиведення (СПН) FVІІІ становить приблизно 12 год у дорослих; період напіввиведення у дітей молодшого віку є коротшим і збільшується з віком. Для розрахунку дози слід помножити масу тіла пацієнта в кілограмах на бажаний рівень FVІІІ (МО/дл), а потім помножити на 0,5.
КФК FІХ доступні з активністю від 250 до 4000 МО. Кожна 1 МО плазмового або рекомбінантного FІХ на 1 кг маси тіла підвищує рівень FІХ у плазмі крові приблизно на 1 МО/дл. СПН FІХ становить приблизно 18-24 год. Немодифіковані рекомбінантні КФК FІХ характеризуються нижчим відновленням фактора в плазмі, ніж плазмові КФК FІХ, тому кожна одиниця FІХ, введена на 1 кг маси тіла, підвищує активність FІХ приблизно на 0,8 МО/дл у дорослих та на 0,7 МО/дл у дітей віком до 15 років [8].
Висока частота інфузій з використанням КФК із СПН збільшує тягар лікування та нерідко призводить до поганого дотримання режимів профілактики пацієнтами. Тому були розроблені продукти з подовженим періодом напіввиведення (ППН) для зменшення кількості ін’єкцій і підвищення рівня просідання FVІІІ(ІХ) для кращого запобігання кровотечам. Для збільшення періоду напіввиведення препаратів для лікування гемофілії успішними виявилися технології злиття молекул і пегілювання. Для продуктів FVІІІ з ППН подовження періоду напіввиведення було обмеженим і не перевищувало відповідне значення для препаратів FVІІІ із СПН у 1,4-1,6 разу (приблизно 19 год). Продукти FІХ з ППН мають набагато довший період напіввиведення, що у 3-5 разів перевищує період напіввиведення таких FІХ із СПН. Пролонгований період напіввиведення для більшості випадків означає можливість введення препаратів 2 рази на тиждень або через кожні 3 дні і 1 раз через кожні 7-14 днів для FІХ [9].
Усі плазмові та рекомбінантні продукти FVІІІ та FІХ зі СПН і ППН, які нині представлені на ринку, наведені в онлайн-реєстрі концентратів факторів згортання крові Всесвітньої федерації гемофілії (ВФГ) [10].
Крім КФК, у значній частині випадків доцільним може бути застосування інших лікарських препаратів. До них належать десмопресин, транексамова кислота, епсилон-амінокапронова кислота [10]. Сьогодні вже створені або розробляються інноваційні терапевтичні засоби з альтернативними способами введення (наприклад, підшкірним) з метою подолати обмеження замісної терапії факторами згортання крові (такі як внутрішньовенне введення, короткий період напіввиведення, ризик утворення інгібіторів).
На сьогодні єдиним ліцензованим препаратом є еміцизумаб. Це гібридне біспецифічне антитіло до ферменту FІХ та зимогену FХ, що імітує кофакторну функцію FVІІІ у пацієнтів з гемофілією А, з інгібіторами або без них. Еміцизумаб зв’язується з FІХ, FІХа, FХ та FХа, однак саме його афінність до FІХа та FХ сприяє активації FХ, що каталізує FІХа й утворення тенази. Ключовими перевагами еміцизумабу є його підшкірне введення, тривалий період напіввиведення, висока ефективність у профілактиці кровотеч та зменшення частоти кровотеч у пацієнтів з інгібіторами FVІІІ або без них. Еміцизумаб не призначений для лікування епізодів гострих кровотеч. Потрібна обережність при лікуванні епізодів проривних кровотеч під час прийому еміцизумабу, оскільки є небезпека розвитку тромбоемболії або тромботичної мікроангіопатії при одночасному застосуванні антиінгібіторного комплексу [11].
Профілактика при гемофілії
Профілактика при гемофілії полягає у регулярному введенні препаратів, спрямованих на підтримку гемостазу, щоб запобігти кровотечам, особливо крововиливам у суглоби, які призводять до артропатії й інвалідності. Профілактика має дати можливість особам з гемофілією вести здоровий та активний спосіб життя, брати участь у більшості фізичних і соціальних видів діяльності на рівні з людьми без гемофілії. Профілактика КФК називається регулярною замісною терапією. Такий режим відрізняється від епізодичної замісної терапії (також відомої як лікування на вимогу), яка визначається як введення КФК лише під час кровотечі. Епізодична терапія, незалежно від дози препарату, хоча і необхідна для зменшення болю й інвалідизуючого впливу окремих крововиливів, істотно не змінює профіль кровотеч, а отже – не змінює природний анамнез гемофілії, що призводить до ураження опорно-рухового апарату й інших ускладнень, зумовлених кровотечами. Тому замість епізодичного лікування рекомендується профілактика. З появою інноваційних нефакторних препаратів замісної терапії, які здебільшого можна вводити підшкірно, профілактика переосмислюється як регулярне введення гемостатичного засобу для посилення гемостазу й ефективного запобігання кровотечам у людей з гемофілією [13].
 Вперше профілактичне лікування запровадили у Швеції в 1950-1960 рр. Мета профілактики полягала у тому, щоб людина з тяжкою формою гемофілії (базовий рівень FVIII(IX) <1 МО/дл) досягла фенотипу кровотеч, властивого для помірної або легкої гемофілії, шляхом постійного підтримання рівня фактора >1 МО/дл. Однак через поступове накопичення нових даних стає дедалі зрозуміліше, що залишкові рівні фактора 1-3 МО/дл є недостатніми для повного уникнення кровотеч у всіх осіб з гемофілією. За таких рівнів періодично відбуваються клінічні та субклінічні кровотечі, що призводить до поступового прогресування захворювання суглобів протягом усього життя [14, 15]. Профілактику характеризують залежно від часу початку та інтенсивності. Ці визначення стосуються і гемофілії А, і гемофілії В (табл. 1, 2).
Вперше профілактичне лікування запровадили у Швеції в 1950-1960 рр. Мета профілактики полягала у тому, щоб людина з тяжкою формою гемофілії (базовий рівень FVIII(IX) <1 МО/дл) досягла фенотипу кровотеч, властивого для помірної або легкої гемофілії, шляхом постійного підтримання рівня фактора >1 МО/дл. Однак через поступове накопичення нових даних стає дедалі зрозуміліше, що залишкові рівні фактора 1-3 МО/дл є недостатніми для повного уникнення кровотеч у всіх осіб з гемофілією. За таких рівнів періодично відбуваються клінічні та субклінічні кровотечі, що призводить до поступового прогресування захворювання суглобів протягом усього життя [14, 15]. Профілактику характеризують залежно від часу початку та інтенсивності. Ці визначення стосуються і гемофілії А, і гемофілії В (табл. 1, 2).

Усі режими профілактики забезпечують значні переваги порівняно з епізодичною терапією. Звичайна профілактика, розпочата на початку життя, асоціюється зі зниженням частоти суглобових кровотеч на 90%, показником середньої річної частоти крововиливів у суглоби нижче 3 на рік і сповільненням погіршення стану суглобів та дегенеративних уражень суглобів. Профілактика також забезпечує захист від інших типів крововиливів при гемофілії, у тому числі запобігає або істотно знижує ризик внутрішньочерепних крововиливів [16]. Довгострокові переваги включають зниження хронічного болю з боку опорно-рухового апарату, функціональних обмежень та інвалідності, зменшення потреби в ортопедичній хірургії, госпіталізаціях, невідкладній допомозі та зменшення перебування у стаціонарі. Через ці переваги Всесвітня організація охорони здоров’я, ВФГ, а також профільні національні й міжнародні організації схвалюють ранню профілактику як стандарт догляду за дітьми з тяжким фенотипом гемофілії протягом усього життя. Крім того, дорослі з тяжким фенотипом гемофілії також повинні розпочати профілактику, якщо вона їм не призначена [2].
Протягом більш ніж двох десятиліть профілактика була стандартом медичної допомоги в більшості країн з розвинутою економікою, але вона рідко проводилась у країнах з обмеженими ресурсами, оскільки вважалася недоступною в стандартних дозах. На початку 2000-х років у низці обсерваційних досліджень було показано переваги низькодозової профілактики КФК (а саме – зменшення епізодів кровотеч і краще збереження здоров’я суглобів) порівняно з епізодичною замісною терапією факторами, яких можна досягти без різкого збільшення фінансових витрат. Тому було визнано, що профілактика КФК у низьких дозах також має бути переважним способом лікування пацієнтів навіть у країнах з обмеженими ресурсами [17].
Інгібітори факторів згортання крові
Інгібіторами при гемофілії називають антитіла IgG до екзогенного FVIII або FIX, які нейтралізують функцію введених КФК. Утворення нового інгібітору слід запідозрити у будь-якого пацієнта з гемофілією, у якого відсутня клінічна відповідь на замісну терапію КФК, особливо у хворих, котрі раніше таку відповідь мали. Контроль над кровотечами є значно більшою проблемою у пацієнтів з інгібіторною формою гемофілії, ніж у пацієнтів без інгібіторів. Інгібітори до FVIII або FIX пов’язані з підвищеною тяжкістю захворювання, включаючи підвищений ризик ускладнень з боку опорно-рухового апарату, болю, фізичних обмежень і проблем із лікуванням, що можуть впливати на фізичне функціонування пацієнта, здатність до фізичних навантажень і якість життя. Наявні істотні відмінності між гемофілією А та гемофілією В щодо частоти виникнення інгібіторів, лікування та відповіді на індукцію імунної толерантності, а також ефективності альтернативних гемостатичних засобів.
Інгібітори визначають за допомогою аналізу Бетезда або Неймеген-модифікованого аналізу Бетезда. Позитивний результат аналізу на інгібітори – це наявність титру >0,6 одиниць Бетезда (ОБ) для FVIII та ≥0,3 ОБ для FIX [1, 4]. Слабкореагуючий інгібітор – це інгібітор з титром <5,0 ОБ, а сильнореагуючий інгібітор – інгібітор з титром ≥5,0 ОБ [5]. Слабкореагуючі інгібітори, як правило, є тимчасовими (транзиторними). Транзиторний інгібітор визначається як позитивний інгібітор, що знижується від зазначеного порогового рівня протягом 6 міс після початкового встановлення без будь-яких змін у схемі лікування та незважаючи на продовження антигенного навантаження КФК. Сильнореагуючі інгібітори зазвичай є стійкими (персистуючими). Їх титр може знижуватися або переставати визначатися після тривалого періоду за відсутності впливу КФК. Однак він збільшується через 3-5 діб після повторного навантаження КФК (анамнестична відповідь).
Інгібітори частіше виявляють при тяжкій формі захворювання, ніж при помірній або легкій, і частіше у пацієнтів з гемофілією А, ніж з гемофілією В. Інші фактори ризику утворення інгібіторів при гемофілії включають наявність інгібіторів у сімейному анамнезі, належність до чорношкірої африканської або латиноамериканської групи, генетичні варіанти, такі як тип мутації гемофілії та поліморфні імунорегуляторні гени, та інтенсивна експозиція КФК (наприклад, інтенсивна замісна терапія КФК при тяжкій кровотечі у ранньому віці, кровотечі в центральну нервову систему, при хірургічному втручанні або травмі). На ризик формування інгібіторів у пацієнтів з гемофілією А може впливати тип препарату (тобто плазмові КФК FVIII з фактором Віллебранда чи без нього або рекомбінантні КФК FVIII). Однак це питання є недостатньо дослідженим і залишається суперечливим [18-23].
Потенційні фактори ризику виникнення інгібіторів:
- раса;
- сімейний анамнез;
- генотип, імунорегуляторні гени;
- тяжкість гемофілії;
- інтенсивність замісної терапії КФК;
- тип КФК.
Нейтралізуючі антитіла формуються з кумулятивною частотою приблизно 30% у раніше не лікованих пацієнтів з гемофілією А. З них 79% виникають протягом перших 20 експозицій, а решта 21% – протягом перших 75 експозицій [24]. Експозиція визначається як будь-який 24-годинний період, протягом якого вводиться препарат, що містить FVIII або FIX. Частота інгібіторної форми у пацієнтів з гемофілією А легкої та середньої тяжкості становить 5-10%, що нижче, ніж у пацієнтів з тяжкою гемофілією. У цьому випадку інгібітори зазвичай виникають у старшому віці та часто після інтенсивної експозиції FVIII, наприклад, при хірургічному втручанні або сильних кровотечах [25]. У більшості випадків ці інгібітори є слабкореагуючими. Інгібітори з сильною реакцією у таких пацієнтів виявляють рідше. Формування інгібіторів у пацієнтів з гемофілією В відбувається з кумулятивною частотою до 5% [26]. Розвиток інгібітору до FIX вважається найсерйознішим ускладненням у пацієнтів з гемофілією В, не тільки через втрату відповіді на замісну терапію FIX, а через й пов’язаний з цим ризик розвитку анафілаксії та нефротичного синдрому. Діти та дорослі зі стійкими інгібіторами до FVIII (ІХ) зазвичай мають вищу частоту госпіталізацій, більші витрати на лікування та вищу смертність, ніж пацієнти без інгібіторів.
Лікування інгібіторної форми включає контроль і запобігання кровотечам та застосування методів усунення інгібіторів [27]. Індукція імунної толерантності (ІІТ) шляхом регулярних і тривалих призначень концентрату FVIII – єдина на сьогодні стратегія з доведеною ефективністю щодо усунення стійких інгібіторів у високому титрі. Високою успішністю (60-80%) характеризуються різні схеми лікування. Незважаючи на більш ніж 40 років клінічного досвіду, оптимальний режим IІT, а також предиктори його ефективності ще обговорюються, тому що відсутні результати великих і методологічно строго витриманих досліджень [28]. Розроблено сучасні критерії для відбору кандидатів і вибір режимів IІT. Згідно з рекомендаціями ВФГ і Британської організації лікарів центрів гемофілії (UKHCDO), сьогодні IІT вже не вважається золотим стандартом з усунення інгібіторів, її слід розглядати як довгострокову інвестицію, в якій високі початкові витрати на лікування окупаються порівняно з вартістю пожиттєвого лікування кровотеч при наявності стійких інгібіторів. Можливо, завдяки профілактиці еміцизумабом, ІІТ можна відтермінувати або уникнути, враховуючи дуже низьку частоту кровотеч у разі застосування цього препарату. Але дискусії щодо цього тривають, а даних недостатньо [29]. Пацієнти з інгібітором FVIII, виявленим більш ніж один раз, який перешкоджає проведенню профілактики або лікування кровотеч при стандартних дозах FVIII, мають пройти IIT для усунення інгібітору та відновлення нормальної клінічної чутливості до FVIII. Оскільки поширеність інгібіторів при гемофілії В низька, досвід використання ІІТ обмежений. Принципи такого лікування подібні до принципів лікування гемофілії А, але рівень успіху є нижчим, особливо у пацієнтів з алергічною реакцією на FIX. В останньому випадку може знадобитися десенсибілізація FIX перед спробою ІІТ, однак дані про ефективність і безпеку такого підходу є недостатніми.
Лікування кровотеч у пацієнтів з гемофілією й інгібіторами необхідно проводити за консультування з центром лікування гемофілії та персоналом, який має відповідний досвід. Препарат для лікування слід обирати з урахуванням титру інгібіторів, клінічної реакції на препарат, місця та характеру кровотечі й наявності препарату в країні [2].
Для слабкореагуючих інгібіторів замісна терапія КФК FVIII при гострих кровотечах є оптимальною, якщо досягаються вимірювані рівні фактора. Необхідний ретельний моніторинг клінічної ефективності, оскільки для досягнення гемостазу можуть знадобитися вищі дози. При сильнореагуючих інгібіторах для лікування кровотеч слід застосовувати обхідні (шунтові) засоби (рекомбінантний активований фактор VIIa – rFVIIa – або концентрат активованого протромбінового комплексу – КАПК, – або свинячий FVIII) [30]. Для пацієнтів із сильнореагуючими інгібіторами, титри яких знизилися до не вимірюваних або низьких, стандартну замісну терапію КФК FVIII можна призначати в екстрених ситуаціях до 3-5 днів (за більш частого введення завдяки коротшому періоду напіввиведення) до виникнення анамнестичної відповіді. За її настання подальше лікування КФК FVIII, як правило, вже неефективне, потрібна терапія обхідними (шунтовими) засобами. Це підкреслює необхідність ретельного моніторингу рівня FVIII (табл. 3).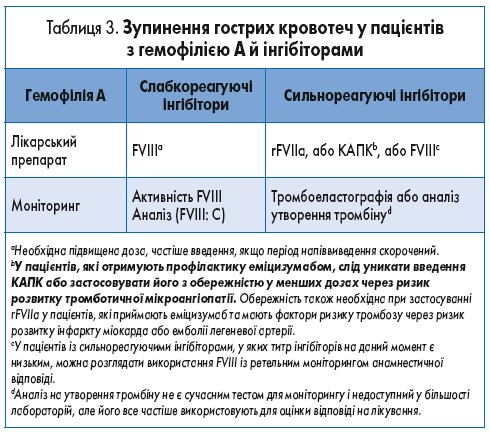
Фактор-замісна терапія – еміцизумаб – все частіше застосовується для запобігання крововиливам у пацієнтів з інгібіторами до FVIII [31]. Введення цього засобу дає змогу ефективно запобігати кровотечам (забезпечує профілактику) у пацієнтів з гемофілією А й інгібітором, але не призначений для лікування кровотеч.
Таким чином, у разі проривних кровотеч необхідне лікування КФК FVIII (при слабкореагуючих інгібіторах), як описано вище, або гемостатичними обхідними засобами (при сильнореагуючих інгібіторах), як описано нижче. Поширені обхідні засоби – rFVIIa та КАПК, які продемонстрували свою ефективність і для профілактики, і для купірування кровотеч [32, 33].
Для пацієнтів з гемофілією В та слабкореагуючими інгібіторами може застосовуватися специфічна замісна терапія КФК FIX, якщо мають місце адекватна нейтралізація інгібітору і надійний контроль кровотечі. Оскільки алергічні реакції та анафілаксія можуть виникати майже у 50% пацієнтів з гемофілією В, у яких є інгібітори, необхідний ретельний контроль такої терапії (табл. 4).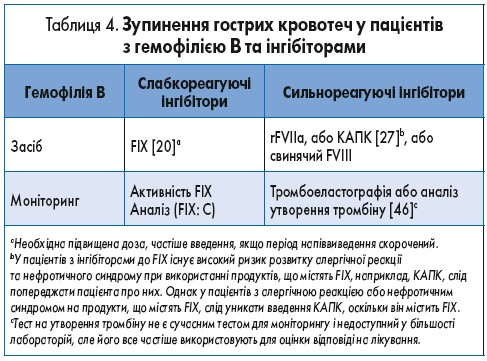
Для пацієнтів з гемофілією В та сильнореагуючими інгібіторами або зі слабкореагуючими інгібіторами, але у яких розвиваються алергічні реакції чи анафілаксія, для контролю кровотечі можна використовувати обхідний засіб rFVIIa. Оскільки КАПК містить FIX, це може спровокувати або посилити алергічну чи анафілактичну відповідь. З цієї причини слід уникати введення КАПК у пацієнтів з гемофілією B. Однак за відсутності такої реакції КАПК забезпечує зіставну з іншими засобами ефективність при контролі гострої кровотечі [33].
На стадії клінічних досліджень перебувають декілька нових нефакторних препаратів для профілактики кровотеч у пацієнтів з гемофілією А та В й інгібіторами, зокрема фітусиран (siRNA-AT3) та анти-TFPI, концентрат rFVIIa із ППН [5, 30, 34]. Ці препарати характеризуються менш інвазивним та/або частим введенням та, якщо підтвердяться їх безпека й ефективність, можуть бути дозволені для клінічного використання.
Список літератури знаходиться в редакції.
Закінчення в наступному номері.
Тематичний номер «Онкологія, Гематологія, Хіміотерапія» № 6 (67) 2020 р.
