


31 березня, 2023
Спадковий рак шлунково-кишкового тракту: клінічні практичні рекомендації ESMO щодо діагностики, лікування та подальшого спостереження
Знання про генетичну схильність до раку шлунково-кишкового тракту постійно поглиблюються з виявленням нових генів. Подібним чином краще розуміння зв’язку генотип/фенотип у пацієнтів із синдромом Лінча (LS) або сімейним аденоматозним поліпозом (CАП) уможливлює надання індивідуалізованих рекомендацій щодо спостереження. Крім того, було показано, що визначення молекулярного профілю хворих на рак може мати вплив на таргетну терапію, таку як імунотерапія. Фахівці, які займаються лікуванням пацієнтів із пухлинами шлунково-кишкового тракту, повинні бути ознайомлені з основними спадковими раковими синдромами та направляти пацієнтів до спеціалізованих онкологічних відділень для належного генетичного консультування та вирішення конкретних проблем, пов’язаних із кожним випадком генетичної схильності.
Синдром спадкового неполіпозного колоректального раку (синдром Лінча)
Поширеність і пенетрантність
LS становить 1-3% усіх діагностованих випадків колоректального раку (КРР). Це спричинено мутаціями зародкової лінії в одному з генів відновлення невідповідності (MMR – MLH1, MSH2, MSH6 або PMS2) чи в молекулі адгезії епітеліальних клітин (EPCAM, яка викликає епігенетичне мовчання MSH2) і успадковується за аутосомно-домінантним типом. Понад 70% мутацій ідентифікуються в MLH1, MSH2 або EPCAM у пухлинах з мікросателітною нестабільністю (MSI).
LS характеризується підвищеним ризиком появи КРР (30-73%) і злоякісних новоутворень інших органів поза ободовою кишкою, таких як ендометрій (30-51%), яєчники (4-15%), шлунок (до 18%), тонка кишка (3-5%), сечові шляхи (2-20%), підшлункова залоза (4%), головний мозок. Носії патогенних варіантів генів MLH1 і MSH2 мають значно вищий ризик розвитку КРР у молодшому віці на момент встановлення діагнозу порівняно з носіями патогенних варіантів MSH6 або PMS2. Кумулятивна захворюваність на рак ендометрія та сечових шляхів вища у носіїв MSH2. Дані про оцінки ризику раку для носіїв EPCAM поки обмежені.
Історично склалося так, що два клінічних фенотипи LS були описані в осіб із зародковими патогенними варіантами гена MMR, які поєднуються із пухлинами центральної нервової системи (синдром Тюрко) або шкірних залоз (синдром Муїра – Торре). Нещодавно третій фенотип, що називається конституційним, або двоалельним дефіцитом MMR (CMMRD), був описаний у тих осіб, які є гомозиготними або складними гетерозиготами для патогенних варіантів гена MMR. Він характеризується плямами на шкірі кольору кави з молоком і пухлинами, що виникають у дитинстві.
Клінічна та молекулярна діагностика
Зміни в генах MMR призводять до накопичення помилок під час реплікації ДНК, особливо в повторюваних послідовностях, відомих як мікросателіти, спричиняє MSI в асоційованих із LS пухлинах. Через зміни в генах MMR у пухлинах LS може бути відсутня експресія відповідного білка MMR (перевірено імуногістохімічним – ІГХ – фарбуванням). ІГХ-тест на MMR у пухлинах КРР має чутливість і специфічність 94 і 88% відповідно і сильно корелює зі статусом MSI.
Клінічні критерії, що використовуються для ідентифікації осіб із ризиком розвитку LS, такі як критерії Amsterdam II і переглянуті рекомендації Bethesda, базуються на віці та сімейному анамнезі раку (табл. 1).

Через обмежену чутливість і специфічність клінічних критеріїв було запропоновано ширший молекулярний ІГХ-скринінг КРР з MMR та/або MSI за допомогою полімеразної ланцюгової реакції (рис.). Було показано, що універсальний ІГХ-тест на MMR пухлини у пацієнтів з КРР є більш чутливим за рекомендації Bethesda для ідентифікації осіб із ризиком LS (100 проти 87,8%). Крім того, клінічні критерії дедалі більше поступаються універсальному ІГХ-тесту на MMR завдяки ролі MSI як біомаркера для прогнозування хорошої відповіді на лікування інгібіторами імунних контрольних точок у пацієнтів із прогресуючим раком. Генетичне тестування зародкових мутацій можна розглядати у пацієнтів, які відповідають критеріям Amsterdam, незалежно від статусу MMR. Якщо доступне мультигенне панельне тестування, гени MUTYH, POLE і POLD1 можна додати до генів MMR, особливо в осіб, у яких пухлину діагностовано у віці до 50 років (клас рекомендацій III; рівень доказовості C).
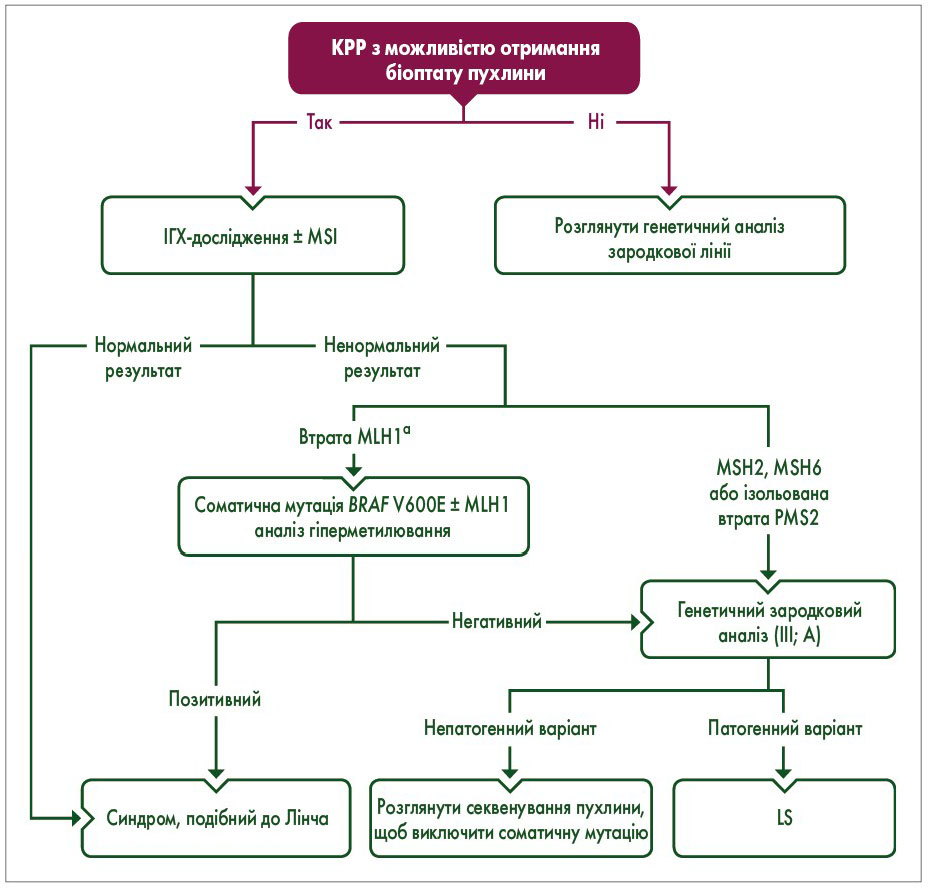 Рис. Алгоритм молекулярної діагностики LS
Рис. Алгоритм молекулярної діагностики LS
a Якщо втрата експресії MLH1 відбувається одночасно з втратою експресії MSH2 або MSH6, слід рекомендувати генетичний аналіз зародкової лінії.
Колоноскопічне спостереження. Була продемонстрована прискорена послідовність аденоми-карциноми в осіб із LS. Періодичне колоноскопічне спостереження дозволяє видаляти поліпи та діагностувати ранню стадію КРР. Колоноскопія, що проводиться через кожні 3 роки, дає змогу знизити захворюваність на КРР і смертність на 62 і 66% відповідно, тоді як частіші скринінгові обстеження асоціюються з ранньою стадією КРР на момент діагностики та зниженням смертності від КРР до 72%. Оскільки діагностика КРР в осіб із LS у віці до 25 років малоймовірна, а ризик КРР у носіїв мутацій генів MSH6 і PMS2 значно нижчий, ніж MLH1 і MSH2, початок колоноскопічного спостереження рекомендується у віці 25 років для носіїв мутацій MLH1 і MSH2 та у віці 35 років для носіїв мутацій MSH6 та PMS2 (III; C). У всіх випадках слід враховувати вік початку захворювання у наймолодшого члена сім’ї та починати спостереження на 5 років раніше (V; B). Оглядова колоноскопія рекомендована через кожні 1-2 роки особам із LS без симптомів (III; A).
Показано, що хромоендоскопія з індигокарміном додатково до стандартної колоноскопії є значно ефективнішою, ніж лише колоноскопія в осіб із LS. Рекомендується проведення високоякісної колоноскопії у спеціалізованих центрах (IV; C).
Спостереження за шлунком і тонкою кишкою. Немає чітких доказів на користь ендоскопічного спостереження верхніх відділів шлунково-кишкового тракту у всіх пацієнтів із LS. Слід розглянути можливість тестування та усунення Helicobacter pylori у всіх носіїв мутації. У регіонах із високим рівнем захворюваності на рак шлунка та в сім’ях з новоутвореннями шлунка в анамнезі можна розглядати можливість ендоскопічного дослідження верхніх відділів травного тракту через кожні 1-3 роки, починаючи з 30-35 років. Рутинне обстеження тонкої кишки при LS характеризується високою частотою хибнопозитивних результатів і не вважається економічно ефективним (IV; C).
Профілактика. Програма профілактики колоректальної аденоми/карциноми 2 (CAPP2) продемонструвала зниження на 60% частоти розвитку КРР та інших пухлин, асоційованих із LS, у тих осіб, які отримували 600 мг ацетилсаліцилової кислоти щодня протягом щонайменше 2 років порівняно з плацебо. Ацетилсаліцилову кислоту можна розглядати як засіб профілактики раку в осіб із LS, хоча оптимальна доза досі не визначена. Це є метою поточного дослідження CAPP3, у якому порівнюють щоденний прийом препарату в дозах 600, 300 і 100 мг (I; C).
Фактори навколишнього середовища та способу життя. Куріння й ожиріння підвищують ризик виникнення аденоми та КРР в осіб із LS. Пацієнтам рекомендується утримуватися від куріння і підтримувати нормальну масу тіла (III; C).
Лікування
Колоректальна хірургія. Показано наявність підвищеного ризику метахронного КРР після часткової колектомії. Якість життя після часткової та повної колектомії з ілеоректальним анастомозом була однаковою. Таким чином, розширена колектомія може бути варіантом лікування пацієнтів з LS, особливо молодшого віку, з приводу КРР (IV; C).
Системне лікування. Наявність MSI є доведеним прогностичним фактором і суперечливим предиктивним фактором для рутинних схем хіміотерапії (ХТ) при КРР та раку шлунка. Сучасні дані не дозволяють дати остаточні рекомендації щодо схем ХТ на основі статусу MMR або MSI. Статус дефіциту MMR або MSI може бути корисним для визначення підгрупи пацієнтів із КРР II стадії, які мають низький ризик рецидиву та для яких ад’ювантна ХТ може бути не потрібна (II; C).
У кількох дослідженнях показано, що MMR-дефіцитні пухлини містять високе мутаційне навантаження й експресують численні неоантигени, це робить їх чутливими до імунотерапії. Два інгібітори імунних контрольних точок забезпечували відповідь у пацієнтів із прогресуючим раком та дефіцитом MMR і отримали прискорене схвалення Управління з контролю якості харчових продуктів і лікарських препаратів США: пембролізумаб для будь-якої солідної пухлини з дефіцитом MMR і ніволумаб для колоректальних пухлин з дефіцитом MMR.
Х-синдром сімейного колоректального раку
Цей синдром наявний у 40% сімей, які відповідають критеріям Amsterdam II спадкового неполіпозного раку товстої кишки, але не мають пухлинного дефіциту MMR або основної альтерації гена MMR зародкової лінії. Ризик розвитку раку в цих сім’ях, імовірно, обмежується пухлинами колоректальної локалізації. Тому рекомендовано колоноскопічний огляд з інтервалом 3-5 років починаючи з 40 років або на 10 років раніше, ніж вік виявлення «наймолодшого випадку» в сім’ї (IV; C).
Синдром дефіциту відновлення конституційної невідповідності
Пацієнти з біалельними мутаціями в одному з генів MMR зазвичай хворіють на рак у дитинстві. Реєструють високу захворюваність на КРР, аденоматозний поліпоз і пухлини тонкої кишки, гематологічні пухлини (лейкоз або лімфома), пухлини головного мозку, ендометрія та сечових шляхів. У двох експертних консенсусах запропоновано спостереження, що включає аналіз крові й ультразвукове дослідження черевної порожнини через кожні 6 місяців, магнітно-резонансну томографію (МРТ) головного мозку, ендоскопічне дослідження верхніх відділів травного тракту та колоноскопію щорічно, а також на розсуд лікаря – щорічно МРТ всього тіла. В обох консенсусах зазначена відсутність надійних доказів і констатована потреба в додаткових дослідженнях.
Синдром, подібний до Лінча (Lynch-like syndrome)
Цей синдром схожий на LS через наявність дефіциту MMR або MSI (за винятком гіперметилювання MLH1), але мутації зародкової лінії відсутні. У цих випадках генетичне тестування MMR на ДНК пухлини дає змогу виявити у 50-70% осіб наявні двоалельні соматичні мутації, якими можна пояснити аномальні результати ІГХ-дослідження та/або MSI. Таким чином, виключення спорадичної соматичної біалельної інактивації цих генів позбавило б інтенсивного спостереження за пухлинами, асоційованими з LS, у родичів, які входять до групи потенційного ризику.
Синдром спадкового поліпозного колоректального раку
Колоректальний поліпоз – це група синдромів, що характеризується множинними поліпами в товстій кишці та підвищеним ризиком КРР, а також позакишковими проявами. Залежно від гістології поліпів виділяють аденоматозний, зубчастий і гамартоматозний поліпоз.
Сімейний аденоматозний поліпоз
Поширеність і пенетрантність
САП є аутосомно-домінантним спадковим розладом, асоційованим із мутаціями зародкової лінії в гені аденоматозної поліпозної палички (adenomatous polyposis coli, APC) і характеризується наявністю кількох колоректальних аденом. При класичній формі пацієнти з САП мають майже 100% ризик розвитку КРР у ранньому віці, якщо не виконати профілактичну колектомію. Це становить <1% усіх випадків КРР і є найчастішою причиною поліпозу з відомою генетичною причиною.
Клінічна та молекулярна діагностика
Клінічний діагноз включає два основні фенотипи: класичну форму, що характеризується значною кількістю (>100) аденом по всій товстій кишці, і форму ослабленого фенотипу, якій притаманні від 10 до 100 аденом, переважно локалізованих у правій половині товстої кишки, та пізніший початок. Це пов’язано з широким спектром пухлин поза ободовою кишкою, включаючи гепатобластому у дітей, рак дванадцятипалої кишки, підшлункової залози, щитоподібної залози та головного мозку. Мутацію зародкової лінії в гені APC виявляють у 80% випадків класичного САП і лише в 10% випадків атенуйованого. Мутації, розташовані між кодонами 1250 і 1464 гена APC, пов’язані з тяжчими формами САП. У 30-40% випадків сімейний анамнез САП відсутній, що свідчить про його походження de novo.
Повне генетичне тестування зародкової лінії має включати як секвенування ДНК, так і великий аналіз перебудови. Аналіз APC повинен включати великі перегрупування (III; A). Завдяки введенню мультигенних панелей генетичне тестування можна проводити як єдиний аналіз кількох генів, залучених до виникнення колоректального аденоматозного поліпозу (APC, MUTYH, POLE, POLD1, NTHL1).
Спостереження та зниження ризику
Спостерігати слід усіх носіїв мутації, а також усіх членів цієї родини, для яких причинну мутацію зародкової лінії не можна ідентифікувати (табл. 2).
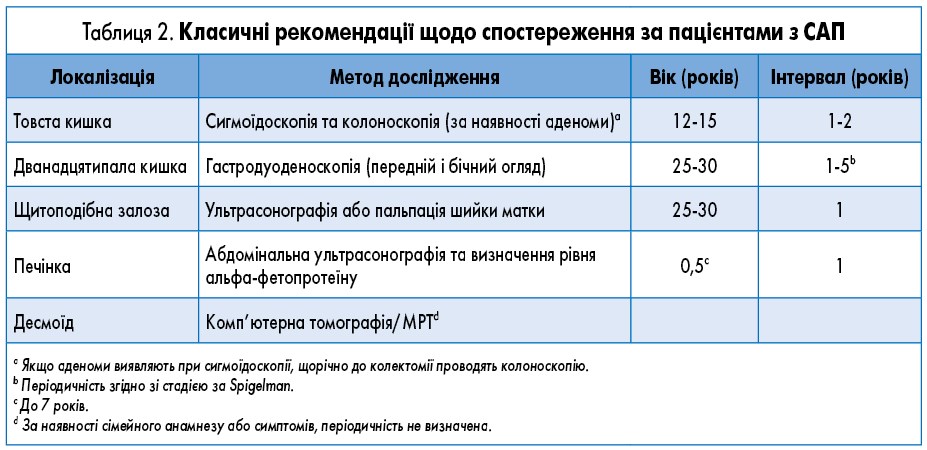
Колоноскопічне спостереження. Пацієнтам із класичним САП починаючи з 12-15 років через кожні 2 роки слід проводити сигмоїдоскопію або колоноскопію. Після виявлення аденоми колоноскопію необхідно виконувати через кожні 1-2 роки до проведення планової колектомії. Пацієнтам із APC-ослабленим САП (AСАП) колоноскопічне дослідження слід виконувати через кожні 1-2 роки починаючи з 18-20 років (III; C).
Лікування класичного САП є хірургічним і має проводитися до 25 років. Вибір хірургічного методу (тотальна колектомія з клубово-прямокишковим анастомозом або проктоколектомія з клубово-анальним анастомозом) залежить від віку на момент встановлення діагнозу, тяжкості поліпозу, наявності поліпів прямої кишки та ризику розвитку десмоїдів. Після хірургічного втручання у пацієнтів із САП рекомендується щорічне або 1 раз на два роки ендоскопічне спостереження (III; B).
Показано, що вторинна профілактика із застосуванням нестероїдних протизапальних препаратів сприяє зменшенню кількості й поширеності колоректальних аденом і, що менш достовірно, аденом дванадцятипалої кишки. Через ризик серцево-судинних подій, асоційованих із прийомом нестероїдних протизапальних препаратів (особливо інгібіторів циклооксигенази-2), жоден із них не схвалений для лікування САП або MUTYH-асоційованого поліпозу (MAP). При їх використанні слід брати до уваги імовірні побічні ефекти (II; B).
Спостереження за шлунком і тонкою кишкою. Поліпи фундальних залоз часто виявляють у пацієнтів з САП, тоді як неоплазії шлунка – рідко. Поліпи дванадцятипалої кишки наявні у близько 90% пацієнтів із САП, а рак дванадцятипалої кишки є другою причиною смерті від раку при САП із сукупним ризиком протягом життя 5%. Таким чином, гастродуоденоскопічне спостереження рекомендується через кожні 5 років починаючи з 25-30 років або після діагностики поліпозу товстої кишки як для пацієнтів із класичним САП, так і з AСАП (III; C). Якщо виявлено аденоми, спостереження ведеться за рекомендаціями Spigelman з урахуванням кількості, розміру та гістологічних характеристик цих новоутворень (табл. 3). У разі I стадії за Spigelman ендоскопічне дослідження верхніх відділів травного тракту рекомендовано через кожні 5 років, а в разі II стадії – через кожні 3 роки, тоді як при більш запущених стадіях інтервали слід скорочувати: через 1-2 роки у разі III стадії і через 6 місяців (або превентивна операція) у разі IV стадії (III; B). Пацієнтам із III і IV стадіями за Spigelman та/або папілярним ураженням рекомендується додатковий ендоскопічний бічний огляд. Аденоми дванадцятипалої кишки зазвичай лікують за допомогою ендоскопічної поліпектомії, хоча в запущених випадках може знадобитися хірургічне втручання (дуоденектомія або дуоденопанкреатектомія). Ризик розвитку раку порожньої та клубової кишок надзвичайно низький, тому рутинне спостереження за допомогою ендоскопічної капсули не рекомендується (V; C).
Позакишкове спостереження. Деякі експерти рекомендують щорічну пальпацію щитоподібної залози та/або ультразвукове дослідження через 2% ризик раку щитоподібної залози протягом життя у пацієнтів із САП. Спостереження щодо гепатобластоми було запропоновано з визначенням рівня альфа-фетопротеїну в сироватці крові та ультразвуковим дослідженням черевної порожнини у дітей пацієнтів із САП 1 раз на 2 роки від народження до 7 років (IV; C). Розвиток десмоїдних пухлин в основному пов’язаний із сімейним анамнезом, операцією на черевній порожнині та місцем мутації; вони можуть виникати всередині живота або в черевній стінці. У цьому випадку необхідно проводити регулярне медичне обстеження та комп’ютерну томографію черевної порожнини або МРТ. Варіанти лікування включають фармакотерапію (нестероїдні протизапальні препарати та/або антиестрогени), хіміотерапію, хірургічне або променеве лікування.
MUTYH-асоційований поліпоз
Поширеність і пенетрантність
MAP є аутосомно-рецесивним синдромом, спричиненим двоалельними мутаціями зародкової лінії в гені MUTYH, і зазвичай характеризується фенотипом ослабленого аденоматозного поліпозу та нижчим ризиком позаободових проявів порівняно з САП. Розвиток поліпів у осіб із двоалельними мутаціями в гені MUTYH зазвичай починається на другому або третьому десятилітті життя. Описано, що ризик розвитку КРР становить 19% у 50 років і 43% у 60 років (середній вік 48 років). Ризик розвитку аденоми дванадцятипалої кишки низький.
Клінічна та молекулярна діагностика
Клінічний спектр мутацій зародкової лінії MUTYH неоднорідний і включає ослаблений та класичний аденоматозний поліпоз, КРР без поліпозу і синдром Лінча. Двоалельні мутації MUTYH слід запідозрити у пацієнтів з ослабленою формою аденоматозного поліпозу або класичним САП із рецесивним типом успадкування. Це також слід припускати у пацієнтів з КРР, який діагностовано до 50 років, і у пацієнтів із множинними поліпами товстої кишки (>10, включаючи як аденоматозні, так і зубчасті).
Найпоширенішими мутаціями в популяції білої раси є Y179C та G396D. Однак наявні етнічні та географічні відмінності в ландшафті мутації цього гена. Поширеність гетерозигот MUTYH у загальній популяції становить 1-2%.
Генетичне тестування має включати всі екзони MUTYH. Через включення мультигенних панелей та істотне перекриття клінічного фенотипу синдромів поліпозу ми рекомендуємо виявлення генів, що беруть участь у виникненні колоректального аденоматозного поліпозу (APC, MUTYH, PELE, POLD1, NTHL1; V; B).
Спостереження та зниження ризику
Колоноскопічне спостереження. Пацієнтам із МАР першу колоноскопію рекомендується проводити у віці 18-20 років і надалі через кожні 1-2 роки (табл. 4). Якщо поліпи неможливо контролювати ендоскопічно, слід розглянути колектомію з клубово-прямокишковим анастомозом за відсутності ураження прямої кишки. Якщо ураження прямої кишки значне – показана тотальна проктоколектомія з клубово-анальним анастомозом. Після операції рекомендується продовжувати спостереження за рештою колоректального сегмента з інтервалом 1-2 роки (III; C).
Скринінг КРР у носіїв моноалельних мутацій рекомендований як для родичів першого ступеня спорідненості пацієнта зі спорадичним КРР. Доказів користі хіміопрофілактики при цьому стані немає.
Спостереження за шлунком і тонкою кишкою. У більшості випадків стратегія спостереження з використанням ендоскопічного дослідження верхніх відділів травного тракту визначається на основі моніторингу поліпів дванадцятипалої кишки, проведення першої ендоскопії у 25-30 років і продовження залежно від стадії за Spigelman (табл. 3).
Поліпоз, асоційований з корекцією полімерази
Недавні дослідження дали змогу ідентифікувати два гени з аутосомно-домінантним успадкуванням, асоційовані із множинними аденомами та раннім розвитком КРР: POLE та POLD1. Мутації в цих генах були пов’язані з різними фенотипами, які варіюють від класичного фенотипу з гастродуоденальним залученням до ослаблених форм або пухлин, характерних LS. Рекомендується підхід як при MAP, із регулярним колоноскопічним спостереженням (табл. 4).
Аденоматозний поліпоз, пов’язаний із зародковою мутацією в NTHL1
У нещодавніх дослідженнях із секвенування цілого екзома виявили асоціацію двоалельної зародкової мутації NTHL1 (16p13.3) з ослабленим аденоматозним поліпозом. Цей новий поліпозний синдром характеризується аутосомно-рецесивним успадкуванням та, ймовірно, підвищеним ризиком розвитку раку ендометрія у носіїв двоалельних мутацій. Немає конкретних настанов щодо ведення цих пацієнтів, тому рекомендується підхід як при MAP, з регулярним колоноскопічним спостереженням.
Синдром зубчастого поліпозу
Поширеність і пенетрантність
Синдром зубчастого поліпозу (SPS) – це стан, що характеризується поєднанням великих та/або численних зубчастих уражень, які поширюються по товстій кишці, з підвищеним ризиком розвитку КРР протягом життя (15-30%). Хоча поширеність SPS не відома, цей синдром стає одним із найчастіших поліпозних синдромів КРР.
Клінічна та молекулярна діагностика
Всесвітня організація охорони здоров’я (ВООЗ) у 2019 році розробила критерії визначення SPS. Критерій 1: ≥5 зубчастих уражень/поліпів проксимальніше прямої кишки, усі мають розмір ≥5 мм, причому ≥2 мають розмір ≥10 мм. Критерій 2: >20 зубчастих уражень/поліпів будь-якого розміру, розподілених по товстій кишці, причому ≥5 розташовані проксимальніше прямої кишки.
Будь-який гістологічний підтип зубчастого ураження/поліпа (гіперпластичний поліп, сидяче зубчасте ураження без або з дисплазією, традиційна зубчаста аденома та некласифікована зубчаста аденома) включається в остаточну кількість поліпів. Кількість поліпів є кумулятивною для кількох колоноскопій. Генетична основа SPS залишається в основному не відомою. Повідомлялося про двоалельні мутації MUTYH у деяких пацієнтів, які відповідають критеріям ВООЗ, зазвичай у контексті супутньої ослабленої форми аденоматозного поліпозу. Нещодавно повідомлялося про мутації зародкової лінії RNF-43 у деяких родинах із SPS.
Спостереження та зниження ризику
Останні дані свідчать про те, що спостереження за допомогою колоноскопії слід проводити через кожні 1-2 роки (це можна продовжити до 2 років у більшості пацієнтів з урахуванням певних факторів ризику, наприклад, множинності поліпів або ознак прогресування; табл. 4; III; C). Хоча потрібні додаткові докази, зазвичай рекомендується проводити колоноскопічний скринінг через кожні 5 років у родичів першого ступеня спорідненості пацієнтів із SPS починаючи з 45 років (або на 10 років раніше, ніж вік встановлення діагнозу наймолодшому хворому члену сім’ї). Немає жодних доказів на підтримку скринінгу з приводу КРР у пацієнтів із SPS. Хірургічне втручання призначають пацієнтам із КРР або тим, кому неможливо безпечно провести ендоскопічне лікування. Тотальна колектомія з клубово-прямокишковим анастомозом є методом вибору у пацієнтів із тяжким і рецидивуючим поліпозом, тоді як сегментарна колектомія може бути показана в менш тяжких випадках. Після колектомії рекомендується продовжувати спостереження за рештою колоректального сегмента з інтервалом 1-2 роки (III; C).
Гамартоматозний поліпоз
Синдроми гамартоматозного поліпозу – Пейтца – Єгерса і ювенільного поліпозу – є рідкісними захворюваннями. Діагностичні критерії та рекомендації щодо спостереження базуються на консенсусній думці експертів (табл. 4; IV; C).
Продовження у наступному номері.
За матеріалами Stjepanovic N. et al. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2019; 30: (10): 1558-1571. Doi: 10.1093/annonc/mdz233.
Підготував Назар Лукавецький
Тематичний номер «Онкологія. Гематологія. Хіміотерапія» № 1 (80) 2023 р.
