


19 липня, 2023
Спадковий рак шлунково-кишкового тракту: клінічні практичні рекомендації ESMO щодо діагностики, лікування та подальшого спостереження (продовження)
Продовження. Початок у № 1 (80), 2023, стор. 14-16.
У більшості випадків рак шлунка (РШ) є спорадичним. Сімейна кластеризація спостерігається приблизно в 10% випадків, а 1-3% є спадковими, охоплюючи спадковий дифузний РШ і сімейний РШ кишкового типу. Також може розвинутися аденокарцинома шлунка та синдром проксимального поліпозу шлунка (GAPPS), який нещодавно визнано рідкісним варіантом САП. Крім того, РШ може розвинутися на тлі інших спадкових ракових синдромів, таких як синдром Лі – Фраумені, САП, Пейтца – Єгерса, LS, спадкового раку грудної залози/яєчника (HBOCS), MAP та ювенільного поліпозу. Ризик виникнення РШ протягом життя при цих синдромах істотно різниться між досліджуваними популяціями, але загалом низький. Алгоритм діагностики спадкового РШ наведено на рисунку.
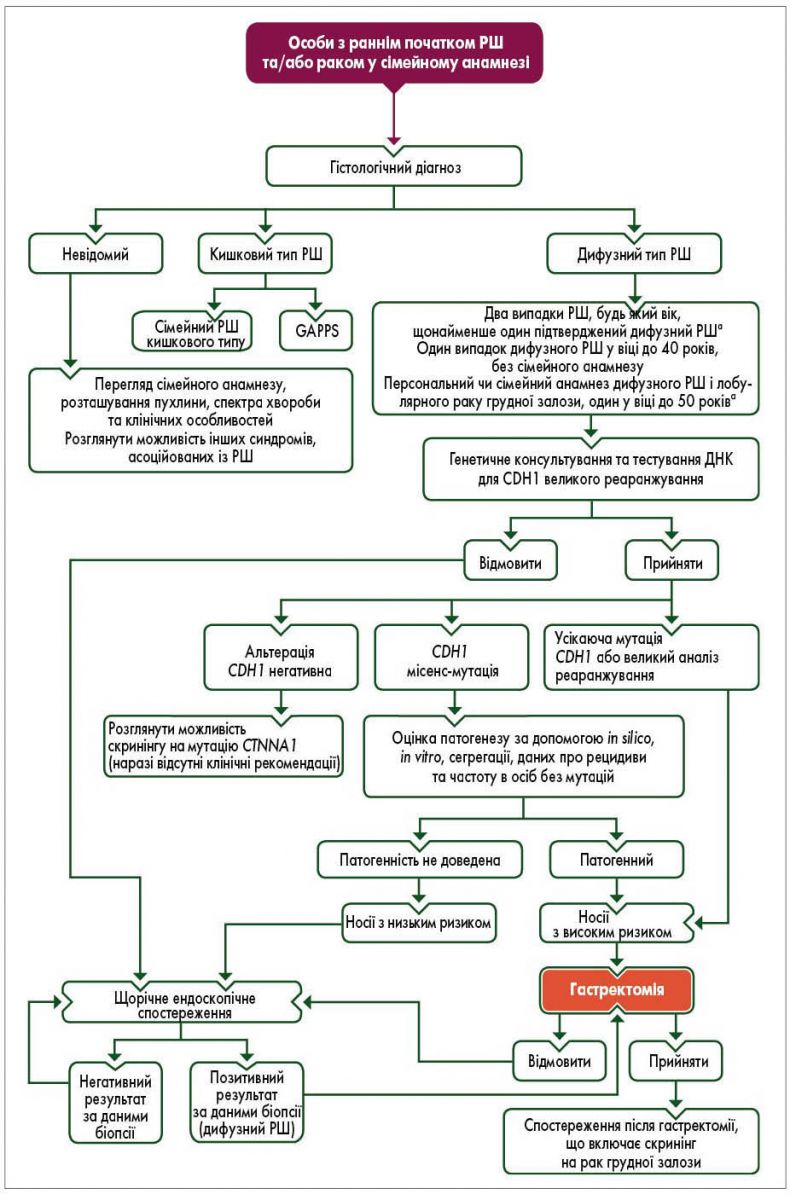 Рис. Алгоритм діагностування спадкового раку шлунка
Рис. Алгоритм діагностування спадкового раку шлунка
а Включаючи родичів першого чи другого ступеня спорідненості.
Спадковий рак шлунка
Спадковий дифузний рак шлунка
Поширеність і пенетрантність. Спадковий дифузний РШ (HDGC) – це аутосомно-домінантний синдром сприйнятливості до раку, що характеризується перснеподібноклітинним раком (SRC)/дифузним РШ (DGC) і часточковим раком грудної залози. Ген CDH1, що кодує E-кадгерин, був ідентифікований як генетична причина HDGC, а нещодавно також був досліджений ген CTNNA1, що кодує α-E-катенін. Частота гетерозиготної мутації зародкової лінії CTNNA1 у сім’ях з DGC низька (~1-2%).
HDGC становить <3% глобальної кількості випадків РШ. Повідомляється, що сукупний ризик розвитку дифузного РШ для носіїв мутації CDH1 у віці до 80 років становить 70% для чоловіків і 56% для жінок. Сукупний ризик виникнення часточкового раку грудної залози для жінок до 80 років з мутацією CDH1 оцінюється в 42%. Вік, у якому формується HDGC, може бути різним – від 14 до 85 років.
Клінічна та молекулярна діагностика. Генетичне тестування на виявлення мутації CDH1 рекомендується в сім’ях з клінічними критеріями HDGC. Тестування зародкових альтерацій CDH1 рекомендується в сім’ях, які відповідають одному з таких трьох критеріїв, запропонованих Міжнародним консорціумом раку шлунка:
- два або більше задокументованих випадки раку шлунка у будь-якому віці у родичів першого або другого ступеня спорідненості з принаймні одним підтвердженим РШ;
- DGC в особистому анамнезі у віці до 40 років або
- DGC та часточковий рак грудної залози в особистому чи сімейному анамнезі (у родичів першого або другого ступеня спорідненості), один діагноз встановлено у віці до 50 років.
Генетичне тестування можна розглядати в сім’ях з випадками двобічного або множинного часточкового раку грудної залози у віці до 50 років, сім’ях з кластеризацією DGC і вовчої губи/вовчого піднебіння та у будь-якого пацієнта з діагнозом in situ або пегетоїдного поширення перснеподібноклітинного РШ.
При визначенні віку, у якому родичі групи ризику мають пройти генетичне тестування, слід брати до уваги наймолодший вік виникнення раку в цій родині. Тестуванню з пізнього підліткового віку або з 20 років життя необхідно надати перевагу в сім’ях із раннім розвитком DGC.
Генетичне зародкове тестування має включати як секвенування ДНК, так і аналіз великої перебудови. Ідентифікація зародкових варіантів місенс-мутацій CDH1 потребує додаткових досліджень для оцінювання їх імовірної патогенності.
Спостереження та зниження ризику
Ендоскопічне спостереження та профілактичне хірургічне втручання. Безсимптомним носіям патогенних мутацій CDH1 пропонується профілактична гастректомія або, для окремих груп, щорічне ендоскопічне спостереження. Спостереження із щорічним ендоскопічним обстеженням рекомендовано для осіб віком <20 років, для тих, хто відмовляється від профілактичної гастректомії, якщо у них немає позитивного результату біопсії, для осіб із супутніми захворюваннями та для тих, хто має сімейний DGC і невідомий варіант CDH1. Рекомендується щонайменше 30 випадкових біопсій під час ендоскопічного обстеження, як описано в Кембриджському протоколі. Будь-яке злоякісне ураження, виявлене ендоскопічно, є показанням до направлення на гастректомію. Однак усі пацієнти, які проходять ендоскопічне обстеження з приводу HDGC, мають бути проінформовані про те, що через вогнищевий і часто ендоскопічно невидимий характер цих уражень цілком можливо, що пухлини не будуть виявлені випадковою біопсією.
Тотальну гастректомію рекомендують у віці від 20 до 30 років. Пацієнтам з позитивним результатом біопсії рекомендована повна резекція шлунка незалежно від віку. Профілактичну гастректомію у віці >75 років слід ретельно розглянути.
Спостереження за раком грудної залози. Щорічна МРТ грудної залози з мамографією з 30 років рекомендується жінкам з мутацією CDH1. Щорічне клінічне обстеження грудної залози і поінформованість пацієнтки та її лікарів про рак грудної залози є важливими.
Сімейний інтестинальний рак шлунка
Поширеність і пенетрантність. Сімейний інтестинальний РШ вважається таким за умови наявності в сімейному анамнезі раку шлунка з аутосомно-домінантним типом успадкування.
Клінічна та молекулярна діагностика. У 1999 році Міжнародний консорціум раку шлунка запропонував діагностичні критерії, аналогічні Амстердамським критеріям у країнах з високим рівнем захворюваності на РШ (наприклад, Португалія, Японія).
Отже, діагностичні критерії в країнах із низьким рівнем захворюваності на РШ включають:
- наявність щонайменше двох родичів першого або другого ступеня спорідненості з РШ інтестинального типу, одному з них діагностовано пухлину до 50 років життя, або
- наявність трьох і більше родичів з інтестинальним типом РШ в будь-якому віці.
Діагноз розглядають, якщо в сімейному анамнезі є РШ кишкового типу в сім’ях без поліпозу. Генетична причина сімейного інтестинального РШ наразі невідома.
Спостереження та зниження ризику. Недостатньо доказів для надання надійних рекомендацій щодо лікування осіб із ризиком сімейного інтестинального РШ. Ерадикація H. pylori рекомендована членам сімей пацієнтів із кишковим типом РШ у віці <40 років або в сім’ях із кластеризацією сімейного інтестинального РШ.
Аденокарцинома шлунка і проксимальний поліпоз шлунка
Поширеність і пенетрантність. GAPPS є аутосомно-домінантним синдромом схильності до раку зі значним ризиком розвитку шлункової, а не колоректальної аденокарциноми.
Клінічна та молекулярна діагностика. Діагностика GAPPS ґрунтується на таких клінічних критеріях:
- поліпи шлунка, обмежені тілом і дном шлунка, без ознак колоректального поліпозу або поліпозу дванадцятипалої кишки;
- >100 поліпів, що вкривають проксимальний відділ шлунка в основному випадку, або >30 поліпів у родичів першого ступеня спорідненості в іншому випадку;
- переважно поліпи фундального відділу шлунка, деякі з ділянками дисплазії (або з диспластичні поліпи фундального відділу шлунка чи аденокарцинома шлунка у родичів);
- аутосомно-домінантний тип успадкування.
Винятки включають інші спадкові синдроми поліпозу шлунка і використання інгібіторів протонної помпи.
GAPPS демонструє неповну пенетрантність. Вік початку РШ варіює (23-75 років; медіана 50 років), а типовий покривний фундальний поліпоз із дисплазією виявляється вже у віці 10 років.
Генетичний дефект був ідентифікований як точкові мутації в промоторі 1B гена APC, які поєднувалися із захворюванням у шести сімействах GAPPS. Тому GAPPS розглядається як варіант САП із переважним шлунковим фенотипом.
Спостереження та зниження ризику. Лікування включає ендоскопічне спостереження з вибірковою біопсією або, бажано, поліпектомією, спрямованою на великі/нерегулярні поліпи, і, зрештою, профілактичну гастректомію. Через обмеженість доступних даних рекомендується індивідуальне лікування.
Спадковий рак підшлункової залози
Поширеність і пенетрантність. Приблизно 10% пацієнтів із раком підшлункової залози (РПЗ) мають рак у сімейному анамнезі. Існує декілька спадкових синдромів, пов’язаних із підвищеним ризиком розвитку РПЗ: HBOCS, сімейна атипова множинна меланома, LS, САП, атаксії-телеангіектазії, Пейтца – Єгерса та спадковий панкреатит. Останні (синдром Пейтца – Єгерса і спадковий панкреатит) пов’язані з найвищим накопиченим ризиком виникнення РПЗ (36 і 18-53% відповідно).
Клінічна та молекулярна діагностика
Діагноз зазвичай ґрунтується на клінічних критеріях різних пов’язаних синдромів з подальшим підтвердженням за допомогою генетичного дослідження. Ці синдроми спадкового раку спричиняють приблизно 10-15% випадків спадкового РПЗ, і найпоширенішою причиною спадкового РПЗ є мутація в гені BRCA2.
У більшості сімей причина спадкового РПЗ не встановлена. Це явище відоме як сімейний РПЗ і стосується сімей з двома або більше родичами першого ступеня спорідненості з PПЗ, які не відповідають критеріям будь-якого іншого синдрому спадкової пухлини. Сімейний РПЗ становить ≤80% кластерів сімей з РПЗ. У деяких останніх дослідженнях повідомляють про зародкові мутації в найбільш «частих» генах (BRCA1, BRCA2, PALB2, CDKN2A), пов’язаних із спадковими панкреатичними синдромами, навіть без інших позапанкреатичних проявів. Це свідчить про те, що у сім’ях із сильною кластеризацією РПЗ доцільним є дослідження панелі мультигенів.
Спостереження за пацієнтами групи високого ризику. Немає переконливих доказів того, що скринінг пов’язаний зі зниженням захворюваності та смертності від РПЗ. На підставі консенсусу Міжнародного консорціуму скринінгу раку підшлункової залози спостереження щодо РПЗ рекомендоване таким пацієнтам групи високого ризику:
- особам з трьома або більше кровними родичами, хворими на РПЗ, з принаймні одним родичем першого ступеня спорідненості, хворим на РПЗ;
- особам із принаймні двома родичами першого ступеня, хворими на РПЗ;
- пацієнтам із синдромом Пейтца – Єгерса незалежно від сімейного анамнезу РПЗ;
- носіям CDKN2A/p16 з одним родичем першого ступеня спорідненості, хворим на РПЗ;
- носіям мутації BRCA2 з одним родичем першого ступеня спорідненості, хворим на РПЗ (або двома хворими членами сім’ї, без родичів першого ступеня спорідненості, хворих на РПЗ);
- носіям мутації PALB2 з одним родичем першого ступеня, хворим на РПЗ;
- носіям мутації MMR (LS) з одним родичем першого ступеня спорідненості, хворим на РПЗ.
На сьогодні щорічне ендоскопічно-ультразвукове дослідження та/або МРТ підшлункової залози є процедурами вибору для спостереження. Програми спостереження зазвичай починають у віці 50 років (або на 10 років раніше, ніж вік наймолодшого хворого родича). Пацієнтам з РПЗ або синдромом Пейтца – Єгерса рекомендується починати спостереження у віці 30 і 40 років відповідно.
Хірургічне лікування пацієнтів з високим ризиком раку підшлункової залози
Не досягнуто консенсусу щодо обсягу резекції підшлункової залози (часткова або повна панкреатектомія) у разі виявлення підозрілого ураження. У цьому випадку необхідне рішення мультидисциплінарної команди, а рішення про хірургічне втручання слід приймати індивідуально. Носіям генної мутації без будь-яких попередніх уражень профілактична панкреатектомія не показана.
Загальні підсумки щодо рекомендацій
Синдром спадкового неполіпозного колоректального раку (синдром Лінча)
- Імуногістохімічне дослідження пухлини на виявлення білків MMR та/або MSI рекомендовано в осіб із КРР [клас рекомендацій III; рівень доказовості A].
- Якщо в пухлині спостерігається втрата MLH1, спочатку слід провести аналіз мутації BRAF V600E або аналіз метилювання промотора MLH1, щоб виключити спорадичний випадок [III; B].
- Пропонується тестування соматичного гена MMR для пацієнтів, у яких на скринінгу виявлено зміни нез’ясованої природи [III; B].
- Клінічний ризик можна оцінити за допомогою Амстердамських критеріїв II або переглянутих рекомендацій Bethesda.
- Імуногістохімічне дослідження на MMR та/або скринінг на MSI з аналізом гіперметилювання промотора MLH1 у випадках втрати експресії MLH1 рекомендоване жінкам із раком ендометрію [III; B].
- Повне генетичне тестування зародкової лінії має включати секвенування ДНК і великий аналіз реаранжування [III; A].
- Рекомендації щодо подальшого спостереження у носіїв мутації включають колоноскопію через кожні 1-2 роки [III; A] та гінекологічний огляд (з трансвагінальним УЗД, визначенням CA-125 та біопсією ендометрію) щорічно у віці від 30 до 35 років [IV; C]. У всіх випадках слід враховувати вік початку захворювання у наймолодшого члена сім’ї та розпочинати спостереження на 5 років раніше [V; B]. Рекомендується проведення високоякісної колоноскопії у спеціалізованих центрах [IV; C]. У пацієнтів з високим ризиком можна розглянути можливість ендоскопічного спостереження верхніх відділів травного тракту (через кожні 1-3 роки, починаючи з 30-35 років). Профілактична гінекологічна операція може бути варіантом для жінок-носіїв, які виношували дитину або перебувають у постменопаузі [IV; C].
Лікування:
- розширена колектомія може бути варіантом у пацієнтів, які проходять первинну операцію з приводу КРР [IV; C];
- статус MMR або MSI можна використовувати для вибору ад’ювантного медикаментозного лікування [II; C];
- при розвинутих пухлинах із дефіцитом MMR можна отримати користь від застосування пембролізумабу або ніволумабу;
- при сімейному Х-синдромі КРР спостереження за допомогою колоноскопії (через кожні 3-5 років) зазвичай слід починати у віці 40 років [IV; C];
- при синдромі Лінча генетичне тестування MMR виключає спорадичні соматичні двоалельні мутації.
Синдроми спадкового поліпозу колоректального раку
- У пацієнтів, у яких виявили >10 колоректальних аденом, слід розглянути можливість панельного генетичного тестування зародкової лінії, яке включає гени APC, MUTYH, POLE, POLD1 і NTHL1. Аналіз APC має включати великі зміни [III; A].
- У сім’ях із класичним САП ректороманоскопію слід починати з 12-15 років і проводити через кожні 1-2 роки. Після виявлення аденоми колоноскопію слід виконувати кожні 1-2 роки до планової колектомії. Хірургічне втручання показане за наявності великої кількості аденом або високого ступеня дисплазії [III; C].
- У сім’ях з AСАП колоноскопію слід проводити через кожні 2 роки, починаючи з 18-20 років і продовжуючи протягом усього життя у носіїв мутації. Хірургічне втручання показане при наявності великої кількості аденом. У деяких пацієнтів із AСАП можливе використання консервативних методів – щорічної колоноскопії та поліпектомії [III; C].
- Тип колоректальної операції при САП (тотальна колектомія з клубово-прямокишковим анастомозом чи проктоколектомія з клубово-анальним анастомозом) залежить від віку пацієнта, тяжкості поліпозу прямої кишки та ризику розвитку десмоїдних пухлин [III; B].
- Після колоректальної операції слід проводити спостереження [III; B].
- Як при класичному САП, так і при AСАП скринінг позакишкових проявів (гастродуоденальний поліпоз, рак щитоподібної залози, десмоїдні пухлини) слід розпочинати після діагностування колоректального поліпозу або у віці 25-30 років, залежно від того, що настане раніше [III; C].
- Якщо виявлено аденоми, слід керуватися класифікацією Spigelman [III; B].
- Аденоми дванадцятипалої кишки зазвичай лікують за допомогою ендоскопічної поліпектомії, хоча хірургічне втручання (дуоденектомія або дуоденальна панкреатектомія) може знадобитися в запущених випадках.
- Пацієнтам, у яких є ризик розвитку десмоїдних пухлин, слід проводити регулярне фізикальне обстеження, комп’ютерну або магнітно-резонансну томографію черевної порожнини й лікування, що включає нестероїдні протизапальні препарати та/або антиестрогени, ХТ, хірургічне видалення або променеву терапію.
MUTYH-асоційований поліпоз
- Біалельні мутації MUTYH слід запідозрити у випадках AСАП або САП із рецесивним типом успадкування, діагностування у віці до 50 років і множинних поліпів товстої кишки.
- Рекомендується мультигенний одноразовий аналіз APC, MUTYH (усі екзони), POLE, POLD1 і NTHL1 [V; B].
- Колоноскопію слід проводити через кожні 1-2 роки з 18-20 років.
- Якщо ендоскопічний контроль неможливий, рекомендується накладання клубово-прямокишкового чи клубово-анального анастомозу, залежно від ступеня ураження прямої кишки, з подальшим щорічним ендоскопічним спостереженням [III; C].
- Рекомендується скринінг на CRC у носіїв моноалельних мутацій.
- Ендоскопічне спостереження верхніх відділів травного тракту за поліпами дванадцятипалої кишки слід починати у віці 25-30 років і продовжувати відповідно до стадії за Spigelman.
Інші синдроми
- Для аденоматозного поліпозу з мутаціями POLE та POLD1, PPAP і NTHL1 колоноскопічне спостереження слід здійснювати за такими самими рекомендаціями, як при MAP.
- При SPS колоноскопічне спостереження слід проводити через кожні 1-2 роки (у більшості пацієнтів його можна продовжити до 2 років залежно від факторів ризику).
- У родичів першого ступеня спорідненості пацієнтів із SPS зазвичай рекомендується колоноскопічний скринінг через кожні 5 років, починаючи з 45 років.
- Пацієнтам із SPS із CRC або при неможливості ендоскопічного лікування показана тотальна колектомія з клубово-прямокишковим анастомозом або сегментарна колектомія з подальшим щорічним спостереженням [III; C].
Спадковий дифузний рак шлунка
- Генетичне тестування на CDH1 рекомендовано в сім’ях із клінічними критеріями спадкового дифузного РШ [III; A].
- Тестування зародкових альтерацій CDH1 рекомендується в сім’ях, які відповідають принаймні одному з критеріїв рекомендацій Міжнародного консорціуму раку шлунка.
- Тестування з пізнього підліткового віку або з 20 років є перевагою в сім’ях з раннім початком дифузного РШ.
- Генетичне тестування зародкової лінії має включати секвенування ДНК і великий аналіз реаранжування.
- Щорічне ендоскопічне спостереження рекомендоване особам віком <20 років, особам, які відмовляються від резекції шлунка, і особам із сімейним дифузним РШ і невизначеним варіантом CDH1.
- Рекомендується мінімум 30 випадкових біопсій [IV; B] і лікувальна гастректомія для пацієнтів з позитивними результатами біопсії, незалежно від віку.
- Тотальну гастректомію рекомендують у віці від 20 до 30 років [IV; A].
- Профілактична гастректомія рекомендована носіям патогенної зародкової мутації CDH1 у віці від 20 до 30 років [IV; A], а щорічне проведення МРТ грудної залози – жінкам-носіям мутації з 30 років [IV; B].
- Важливе значення має щорічне клінічне обстеження грудної залози й поінформованість пацієнтки та її лікарів про рак грудної залози.
Сімейний рак шлунка кишкового типу
- Діагноз сімейного РШ кишкового типу розглядається, коли в анамнезі сімей без поліпозу є РШ кишкового типу.
- Неможливо дати надійні рекомендації щодо ведення осіб групи ризику [V; C], але ерадикація Helicobacter pylori рекомендована членам сімей пацієнтів із РШ кишкового типу віком <40 років або сім’ям із кластеризацією сімейного РШ кишкового типу.
Аденокарцинома шлунка і проксимальний поліпоз шлунка
- GAPPS діагностується відповідно до ступеня та поширення поліпів шлунка й сімейного анамнезу.
- Лікування має бути індивідуальним і включати ендоскопічне спостереження з випадковими біопсіями або поліпектоміями та можливою профілактичною гастректомією.
Спадковий рак підшлункової залози
- Мультигенне панельне тестування, що охоплює BRCA1, BRCA2, PALB2, CDKN2A, рекомендоване сім’ям із сильною кластеризацією РПЗ [IV; B].
- Спостереження, як правило, починають у віці 50 років (або на 10 років раніше, ніж вік наймолодшого ураженого родича) [IV; B], а щорічне ендоскопічне УЗД та/або МРТ підшлункової залози є процедурами вибору.
- Пацієнтам зі спадковим панкреатитом або синдромом Пейтца – Єгерса рекомендується починати спостереження у віці 30 і 40 років відповідно.
- При підозрілих ураженнях хірургічне втручання має плануватися індивідуально.
- Профілактична панкреатектомія не показана носіям генної мутації без будь-яких передракових уражень [V; A].
За матеріалами Stjepanovic N. et al. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2019; 30: (10): 1558-1571. Doi: 10.1093/annonc/mdz233.
Підготував Назар Лукавецький
Тематичний номер «Онкологія. Гематологія. Хіміотерапія» № 3 (82) 2023 р.
