


19 липня, 2023
Церебральна хвороба дрібних судин: механізми виникнення, клінічні прояви, діагностика та ведення пацієнтів
Частина 1 Частина 2 Частина 3
За допомогою шкали Fazekas оцінюють ушкодження білої речовини, при цьому краще використовувати режим FLAIR, де 0 – немає лейкоареозу, 1 – множинні точкові ураження, 2 – помірний зливний лейкоареоз, 3 – тяжкий «зливний» лейкоареоз (рис. 5). Шкала розподіляє білу речовину на перивентрикулярну та глибоку білу речовину; кожній ділянці надається ступінь залежно від розміру й злиття уражень. Перивентрикулярна біла речовина: 0 – відсутній, 1 – «шапки» чи тонкий прошарок, 2 – гладкий «ореол», 3 – нерегулярний перивентрикулярний сигнал, що поширюється в глибоку білу речовину. Глибока біла речовина: 0 – відсутній, 1 – точкові вогнища, 2 – початок злиття, 3 – великі зони злиття [9].
![Рис. 5. Зміни білої речовини (лейкоареоз), оцінені за шкалою Fazekas (модифіковано згідно з Qu M. et al., 2020) [39]](/multimedia/userfiles/images/2023/ZU_11_2023/ZU_11_2023_pic_5_st_16_17_.jpg)
Рис. 5. Зміни білої речовини (лейкоареоз), оцінені за шкалою Fazekas (модифіковано згідно з Qu M. et al., 2020) [39]
Важливо, що етіологія змін перивентрикулярної білої речовини та глибокої (субкортикальної) білої речовини відрізняється. Остання має хронічну ішемію дрібних судин за своєю природою, тоді як перша (перивентрикулярна) пов’язана з поєднанням демієлінізації, гранулярного епендиміту та субепендимального гліозу, а також ішемії дрібних судин [21].
Нещодавно було запропоновано термін «загальний бал ЦХДС», який включає 4 тісно корельовані ознаки, котрі є маркерами ЦХДС (гіперінтенсивність білої речовини, лакуни, видимі периваскулярні простори, церебральні мікрокрововиливи). Загальна оцінка ЦХДС може забезпечити повнішу оцінку впливу ЦХДС на мозок простим і прагматичним способом. Вплив ЦХДС візуально оцінюється за шкалою від 0 до 4, де 1 бал визначає наявність однієї з 4 ознак ЦХДС (табл. 3) [5, 42].
|
Таблиця 3. Характеристики та категорії загальної оцінки захворювань дрібних судин (модифіковано згідно зі Staals J. et al., 2014) [42] |
||
|
МРТ-ознака |
Дефініція |
Шкала |
|
Лакуни |
≥1 лакуна |
1 бал |
|
Крововиливи |
≥1 крововилив |
1 бал |
|
Периваскулярний простір |
Розширення периваскулярних просторів у базальних гангліях від середнього до тяжкого |
1 бал |
|
Гіперінтенсивність білої речовини |
Перивентрикулярна гіперінтенсивність білої речовини Fazekas 2-3 |
1 бал |
Установлено, що фактори ризику ЦХДС – куріння, гіпертонія, вік і чоловіча стать – пов’язані з загальною сумарною оцінкою ЦХДС [42].
Окрім візуалізаційних методів дослідження, слід провести детальне фізикальне обстеження нервової системи, яке має передбачати оцінку когнітивних функцій за допомогою відомих шкал (МоСА, VaDAS-Cog), дисфункцію сфінктера, супутні порушення ходи та псевдобульбарного паралічу.
На відміну від нейродегенеративних патологій, як-от хвороба Альцгеймера, рівні Aβ40, Aβ42, а також T-tau, P-tau в спинномозковій рідині в більшості пацієнтів із когнітивними порушеннями в разі ЦХДС є нормальними.
Тести на мутантні гени можуть допомогти в етіологічному діагнозі когнітивних порушень при ЦХДС. Наприклад, мутація гена Notch 3 є основою церебральної автосомно-домінантної артеріопатії з підкірковими інфарктами, лейкоенцефалопатією (CADASIL) [37].
У дослідженні D. Yao та співавт. (2022) виявили можливий зв’язок між цистатином С і ЦХДС, особливо для лакун, що не залежало від оціненої швидкості клубочкової фільтрації. Цистатин С – потужний інгібітор цистеїнової протеази; відіграє плейотропну роль у патофізіології судин людини та є маркером функції нирок, а також може пригнічувати активність цистеїнових протеаз, у т. ч. катепсину B, L, H, і вважається основним ендогенним інгібітором цистеїну катепсинів. Дисбаланс між цистатином С і експресією цистеїну катепсинів може зумовити ремоделювання судинної стінки та нейрозапалення, які є поширеними патологічними процесами в дрібних судинах головного мозку. Нові докази показали, що циркулювальний цистатин С мав сильний зв’язок із ризиком ішемії та інсульту, може бути пов’язаний з церебральними маркерами візуалізації ЦХДС (гіперінтенсивність білої речовини, церебральні мікрокровотечі) в популяції [52].
Диференційна діагностика
Зміни, схожі на ЦХДС, можуть з’являтися за значної кількості патологій ЦНС, однак здебільшого необхідно диференціювати ЦХДС із патологіями, представленими в таблиці 4.
|
Таблиця 4. Диференційна діагностика ЦХДС [13, 32, 35] |
|
|
Патологія |
Дані, які свідчать на користь діагнозу |
|
Мультифокальна прогресувальна лейкоенцефалопатія |
- за наявності лейкоенцефалопатії відсутні церебральні лакуни; - вогнища на МРТ частіше локалізовані на з’єднанні сіро-білої речовини з ушкодженням юкстакортикальних асоціативних волокон; - при клінічному огляді часто спостерігаються дефіцит поля зору та порушення мови, а також порушення координації рухів і ходи; - дані, які свідчать про наявність імунодефіциту (опортуністичні інфекції, лімфопроліферативні захворювання, імуносупресивна терапія); - виявлення JC-вірусу в лікворі |
|
Токсична лейкоенцефалопатія хронічного впливу |
- з’являється на тлі впливу лейкотоксичних речовин (опіоїди, кокаїн, амфетаміни, героїн, толуол тощо); - проявляється на МРТ гіперінтенсивністю білої речовини, яка є симетричною, особливо зі змінами кортикоспинальних шляхів моста, перироландової білої речовини, білої речовини мозочка зі збереженням юкстакортикальної білої речовини та сірої речовини; - наявні зміни в інших органах і системах, зумовлені хронічним токсичним впливом відповідних речовин |
|
СНІД-асоційована енцефалопатія / деменція |
- проявляється симетричним перивентрикулярним типом змін білої речовини зі збереженням юкстакортикальної та інфратенторіальної білої речовини, атрофією сірої речовини кори головного мозку, атрофією глибокої білої речовини й об’ємними змінами базальних ганглій; - позитивні тести на СНІД |
|
Розсіяний склероз |
- ураження зорового нерва та спинного мозку; - накопичення вогнищами контрасту; - ураження мозолистого тіла (пальцеві втиснення Доусона); - зміни ліквору, інтратекальний синтез IgG, виявлений олігоклональними смугами; - нормальний рівень піровиноградної та молочної кислот плазми й ліквору, відсутність мутацій мітохондріальних ДНК, змін біопсії скелетних м’язів (при диференціації з MELAS); - відсутність автоантитіл до системних хвороб і змін із боку крові (зміни ШОЕ, рівні СРБ); - молодий вік та відсутність судинних факторів ризику |
Характеристика окремих типів ЦХДС
І тип: артеріолосклероз (або пов’язана з віком і судинними факторами ризику ЦХДС)
Для опису цього типу широко використовується термін «гіпертонічна артеріопатія» – форма ЦХДС, часто пов’язана з багатьма судинними факторами ризику, як-от гіпертонія, діабет тощо. Однак термін «гіпертонічна артеріопатія» дещо вводить в оману, адже це захворювання не завжди обов’язково пов’язане саме з гіпертонією (з урахуванням інших факторів ризику). Цю форму ЦХДС також часто називають артеріолосклерозом (віковим або судинним) [5].
Інший термін, яким описується ця патологія, – хвороба Бінсвангера. Термін «хвороба Бінсвангера» запровадив Алоїс Альцгеймер у 1902 році на честь свого професора Отто Бінсвангера, який уперше описав клінічні та патологічні аспекти захворювання в 1884 році, а через 60 років Ольшевський запровадив термін «підкіркова артеріосклеротична енцефалопатія» [45].
Серед цих форм ЦХДС найчастіше зустрічаються артеріолосклероз і церебральний малий атеросклероз судин. Артеріолосклероз, судинний фактор ризику ЦХДС, як відомо, пов’язаний з віком і є найпоширенішою зміною дрібних судин у літньому мозку. Вираженість артеріолосклерозу посилюється з віком і загострюється гіпертонічною хворобою та цукровим діабетом (рис. 6) [27].
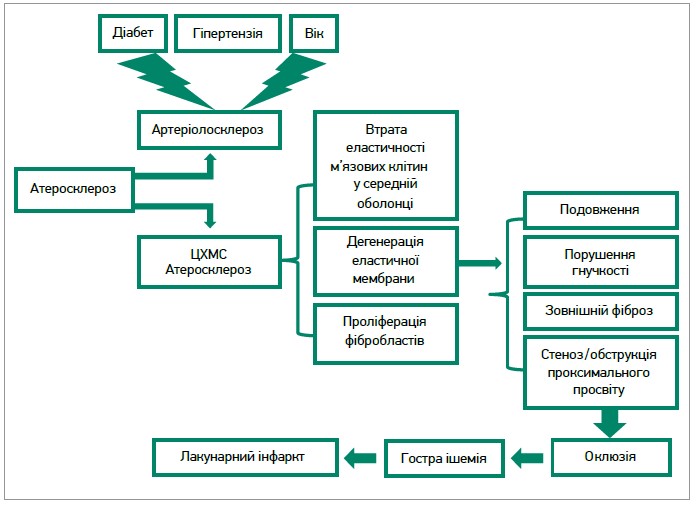
Рис. 6. Розвиток ЦХДС під впливом різних факторів ризику (модифіковано згідно з Li Q. et al., 2018)
Ураження за ЦХДС пов’язані з судинними факторами ризику, але ЦХДС здебільшого не є атероматозною або тромбоемболічною оклюзійною хворобою. Може з’явитися звуження просвіту мікросудин із тромботичною оклюзією, отже, ішемія виникає вторинно та сама собою не є першопричиною зміни ендотелію стінки судин, периваскулярної або білої речовини. Більшість спорадичних ЦХДС – наслідок вродженої чи набутої церебральної ендотеліальної дисфункції, що проявляється у вигляді порушення вазореактивності, проникнення ГЕБ, зміненої пульсації артеріальної, венозної та спинномозкової рідини, порушення глімфатичного транспорту. Кілька факторів у ранньому віці, включаючи когнітивні здібності, рівень освіти, соціально-економічні негаразди, збільшують ризик ЦХДС у подальшому житті, а це свідчить про те, що ЦХДС виникає не лише через вплив судинного фактора ризику всередині чи наприкінці життя, а може мати початок у молодшому віці. Дисфункція ендотеліальних клітин наявна в неонатальних моделях щурів зі спорадичною як у людей ЦХДС, яка виникає задовго до впливу факторів ризику та перешкоджає клітинам-попередникам олігодендроцитів дозріванню й утворенню мієліну, сприяючи уразливості білої речовини в старшому віці [50].
Окремо визначається термін «прихована» ЦХДС (пЦХДС), яка часто зустрічається на нейровізуалізації в осіб без явних неврологічних проявів, що підвищує ризик майбутнього інсульту, когнітивних розладів і смерті [49].
На КТ хвороба представлена змінами підкіркового відділу у вигляді двобічного ураження білої речовини низької щільності. Ці ураження найчастіше наявні в перивентрикулярних ділянках, особливо прилягають до лобових рогів. Юкстакортикальна біла речовина (U-асоціативні волокна) часто зберігається. Порушення білої речовини краще характеризуються на МРТ, яка має більшу чутливість, ніж КТ. Ураження зазвичай є двобічними, симетричними, групуються навколо передніх рогів, можуть бути наявні в білій речовині стовбура головного мозку, особливо в центральному мості. Також можуть спостерігатися підкіркові мікрокрововиливи, котрі можна виявити на послідовностях SWI, але їхня наявність у значній кількості (чи якщо вони розташовані в кортикальних ділянках) спричиняє появу підозри на амілоїдну ангіопатію [45].
ІІ тип: ЦАА
ЦАА є переважно спорадичною патологією, значно пов’язаною з віком і хворобою Альцгеймера. Захворювання рідко зустрічається у віці до 50 років [20].
ЦAA – цереброваскулярне захворювання, зумовлене відкладенням β-амілоїду (Aβ) в стінках артерій, артеріол, капілярів головного мозку. Депонований матеріал складається із продуктів розпаду білка-попередника амілоїду, який розщеплюється β- і γ-секретазами на Aβ із фрагментами різної довжини амінокислот (Aβ40, Aβ42) [8, 27].
Aβ у разі ЦAA є переважно розчинною 40-амінокислотною структурою (Aβ1-40), тоді як Aβ у сенільних бляшках при хворобі Альцгеймера – це довша та менш розчинна 42-амінокислотна структура (Aβ1-42). Ці амілоїдні білки походять від білка-попередника амілоїду (АРР), кодованого в гені на 21,5 хромосомі; продукуються в багатьох ділянках тіла, але експресуються й метаболізуються в Aβ в особливо високих рівнях нейронами мозку. Через цитотоксичну дію Aβ виникає дегенерація, а також спостерігається остаточна втрата гладкої мускулатури, що зумовлює стоншення та ламкість стінки судини [43].
ЦAA вибірково залучає судини головного мозку (насамперед, лептоменінгеальні та кортикальні). Відкладення Aβ спочатку відбувається в середній оболонці й адвентиції, пізніше Aβ накопичується в усіх шарах стінки судини та спричиняє втрату клітин гладкої мускулатури. Згодом відбувається порушення кровообігу в стінці судини, що зумовлює утворення мікроаневризм і фібриноїдного некрозу [8].
У разі ЦАА ураження судин проявляється характерною плямистістю, завдяки чому його вогнища можуть знаходитися поряд із нормальними незміненими судинами. ЦAA також має схильність локалізуватися біля потиличної ділянки, адже потиличні судини за своєю природою є товстішими та можуть умістити більше відкладеннь Aβ порівняно із судинами інших відділів головного мозку [43].
Трьома кардинальними біомаркерами нейровізуалізації є лобарний ВМК, нетравматичний конвекситальний субарахноїдальний крововилив (кСАК) / кортикальний поверхневий сидероз і лобарні церебральні мікрокровотечі [20].
Спонтанний ВМК є найбільш інвалідизувальним проявом розриву тонких і крихких Aβ-навантажених судин при ЦAA та становить до 74% усіх спонтанних ВМК у літніх людей. Через Aβ відкладення зі схильністю до кортикальних та лептоменінгеальних судин, крововиливи, пов’язані з ЦAA, мають тенденцію виникати в периферичних кіркових і підкіркових «лобарних» місцях. Характерна лобарна локалізація ВМК, пов’язаного з ЦAA, є важливою, оскільки це може бути ключовим фактором диференціації причини ВМК у «глибоких» місцях за таких патологій, як хронічна гіпертензивна артеріопатія, яка має тенденцію до переважного ураження базальних ганглій і мосту в літніх людей. Клінічні прояви лобарного ВМК варіюють залежно від розташування та розміру крововиливу, але включають головний біль, фокальний неврологічний дефіцит, фокальні судоми, нудоту і блювання, пригнічений стан свідомості. Діагноз ЦAA до прояву ВМК має першочергове значення, особливо з огляду на високу поширеність використання антиагрегантів і антикоагулянтів у літніх пацієнтів для профілактики інших супутніх захворюваннь, які можуть збільшити ризик цього наслідку [43].
До нетравматичного кСАК і кортикального поверхневого сидерозу належать нетравматичні крововиливи в ≥1 кортикальних борознах без розповсюдження на базальні цистерни, сильвієву / міжпівкульову щілину чи шлуночки, що спричиняється витіканням крові з сильно ураженої крихкої поверхневої судини (рис. 7). Прогресує кСАК до кіркового поверхневого сидерозу – криволінійні гіпоінтенсивні формування за кортикальною поверхнею, видимі на чутливій до крові послідовності магнітного поля МРТ [20].
![Рис. 7. а) поверхневий сидероз у правій півкулі (хронічний гемосидерин); б) Т2-зважене зображення, що демонструє кСАК правої півкулі внаслідок ЦAA; в) численні лобарні церебральні мікрокровотечі, що відповідають ЦAA (чорна стрілка) (модифіковано згідно з DeSimone C.V. et al., 2017) [8]](/multimedia/userfiles/images/2023/ZU_11_2023/ZU_11_2023_pic_7_st_16_17.jpg)
Рис. 7. а) поверхневий сидероз у правій півкулі (хронічний гемосидерин); б) Т2-зважене зображення, що демонструє кСАК правої півкулі внаслідок ЦAA; в) численні лобарні церебральні мікрокровотечі, що відповідають ЦAA (чорна стрілка) (модифіковано згідно з DeSimone C.V. et al., 2017) [8]
Хоча кСАК може проявлятися головним болем за типом «удар грому», класично головний біль за кСАК зазвичай є дуже слабким або не виникає узагалі. Натомість кСАК проявляється розвитком транзиторних фокальних неврологічних симптомів. Транзиторні фокальні неврологічні симптоми мають вигляд повторюваних стереотипних парестезій, які тривають декілька хвилин, коли кСАК локалізується на центральній борозні, що знаходиться біля первинної моторної та сенсорної кори, а також візуальних симптомів, парезів, дисфагії [43].
Церебральні мікрокровотечі – це точкові гіпоінтенсивні утворення, що спостерігаються на чутливій до крові послідовності магнітного поля МРТ, які являють собою екстравазацію крові з накопиченням гемосидерину в макрофагах [20]. Більшість церебральних мікрокровотеч мають діаметр від 2 до 5 мм або (рідко) до 10 мм [40].
Існує низка негеморагічних знахідок на візуалізації, які також свідчать про діагноз ЦAA, до яких належать гіперінтенсивність білої речовини, розширення периваскулярних просторів, лобарні лакуни та кортикальні мікроінфаркти [22].
Діагностика ЦАА проводиться з використанням модифікованих Бостонських критеріїв, за яких визначається достовірна, ймовірна та можлива ЦАА (табл. 5)
|
Таблиця. 5. Модифіковані Бостонські критерії діагнозу ЦАА (модифіковано згідно з Rost N.S. і Etherton M., 2020) [40] |
|||
|
Наявна патологія |
Немає патології |
||
|
достовірна |
імовірна ЦАА |
імовірна ЦАА |
можлива ЦАА |
|
посмертне дослідження: → лобарний внутрішньопаренхіматозний крововилив; → тяжка ЦАА з васкулопатією |
патологічна тканина: → ЦАА в зразку (біопсія або евакуйована гематома) |
|
|
|
|
дані візуалізації: → лобарний інтрапаренхіматозний крововилив, церебральні мікрокровотечі чи кортикальний поверхневий сидероз |
дані → множинні лобарні внутрішньопаренхіматозні крововиливи чи церебральні мікрокрововиливи; → поодинокі лобарні інтрапаренхіматозні крововиливи та кортикальний поверхневий сидероз |
дані візуалізації: → поодинокий лобарний інтрапаренхіматозний крововилив, церебральні мікрокровотечі чи кортикальний поверхневий сидероз |
III тип: спадкова чи генетична ЦХДС
Моногенні розлади становлять від ≈1 до 5% усіх випадків інсульту. Найпоширенішими моногенними захворюваннями, пов’язаними з розвитком ЦХДС та інсультами, є церебральна автосомно-домінантна артеріопатія з підкірковими інфарктами, лейкоенцефалопатією (CADASIL), ХФ, мітохондріальна міопатія, енцефалопатія, лактат-ацидоз з інсультоподібними епізодами (MELAS) та такими, як синдром COL4A1, церебральна автосомно-рецесивна артеріопатія з підкірковими інфарктами, лейкоенцефалопатією (CARASIL), спадкова ендотеліопатія з ретинопатією, нефропатією, інсультами (HERNS) [3, 40].
Моногенні захворювання зустрічаються рідко, але відіграють значну роль у розвитку інсультів, особливо в молоді. Генетичний стан, пов’язаний з інсультами, може бути заснованим на мутації одного гена чи кластера генів з автосомно-домінантним або рецесивним успадкуванням. Клінічні ознаки генетичних захворювань, пов’язаних з інсультами, мають широкий спектр симптомів. Діагноз часто є дуже складним через збіг фенотипів між розладами та гетерогенністю фенотипів у родинах [3].
Основні спадкові патології ЦХДС (сЦХДС) та їхні візуалізаційні / клінічні характеристики представлено в таблиці 6.
|
Таблиця 6. Моногенні захворювання, пов’язані зі спадковою ЦХМС (модифіковано згідно з Rost N.S. і Etherton M. et al., 2020) [40] |
|||
|
Патологія |
Генетична основа / успадкування |
Зміни на МРТ |
Клінічна маніфестація |
|
CADASIL (церебральна автосомно-домінантна артеріопатія з підкірковими інфарктами, лейкоенцефалопатією) |
NOTCH3 (автосомно-домінантне, AД) |
Поширена гіперінтенсивність білої речовини з переважанням у скроневих ділянках, церебральні мікрокровотечі, атрофія мозку |
Рецидивувальні інсульти, ТІА, деменція, депресія, мігрень з аурою. Вік початку зазвичай складає |
|
CARASIL (церебральна автосомно-рецесивна артеріопатія з підкірковими інфарктами, лейкоенцефалопатією) |
HTRA1 (автосомно-рецесивний, AР) |
Поширена гіперінтенсивність білої речовини, атрофія мозку |
Рецидивувальні інсульти, ТІА. |
|
Сімейна ЦАА |
АРР (проамілоїдний білок), CST3 (цистатин С), ITM2B (білок інтегральної мембрани 2B), TTR (транстиретин) (AД) |
Поширена гіперінтенсивність білої речовини, атрофія мозку, лобарні церебральні мікрокровотечі, кірковий поверхневий сидероз |
Лобарні інтрапаренхіматозні крововиливи, деменція, судоми. Вік початку захворювання складає між 45 і 65 роками |
|
HANAC (спадкова ангіопатія з нефропатією, аневризмами, м’язовими судомами) |
COL4A1, COL4A2 (колаген типу IV А) (сімейний, спорадичний) |
Поширена гіперінтенсивність білої речовини, церебральні мікрокровотечі, внутрішньочерепні аневризми |
Звивистість артеріол сітківки, крововиливи, неонатальні інсульти, поренцефалія, внутрішньопаренхіматозний крововилив, нефропатія |
|
ХФ |
GLA (α-галактозидаза) (Х-зв’язане) |
Гіперінтенсивність білої речовини |
Інсульти дрібних судин, деменція, невропатія дрібних волокон, ангіокератоми, ниркова та серцева недостатність |
|
Гомоцистинурія |
CBS (цистатіонін-β-синтаза) (AР) |
Гіперінтенсивність білої речовини |
Інсульт, передчасний атеросклероз, вивих кришталика, марфаноподібні ознаки, когнітивні порушення та розумова відсталість |
|
MELAS (мітохондріальна енцефаломіопатія, лактат ацидоз та інсультоподібні епізоди) |
Множинні мітохондріальні гени (по материнській лінії) |
Гіперінтенсивність білої речовини |
Інсульт, мітохондріальна міопатія, лактоацидоз, затримка розвитку, сенсоневральна втрата слуху, судоми |
|
HDLS (спадкова дифузна лейкоенцефалопатія зі сфероїдами) |
CSF1R (рецептор колонієстимулювального фактора 1) (AД) |
Значна гіперінтенсивність білої речовини |
Деменція, зміни особистості та поведінки, судоми, паркінсонізм |
|
RVCL (васкулопатія сітківки із церебральною лейкодистрофією |
TREX1 (AД) |
Значна гіперінтенсивність білої речовини |
Інсульт, прогресувальна втрата зору, нефропатія |
|
Синдром Аксенфельда – Рігера |
FOXC1, PITX2 (AД) |
Гіперінтенсивність білої речовини, підкіркові інфаркти |
Інсульт, звивистість артеріол сітківки, вади розвитку мозочка |
Під час діагностики сЦХДС з’являються певні труднощі через схожість змін при нейровізуалізації та клінічних проявів інших типів ЦХДС, тому наявність червоних прапорців, які можуть спричинити підозру на сЦХДС, є причиною для того, щоб схилити клініциста до детальнішого дообстеження хворого, включаючи визначення генетичних маркерів захворювання, а це може вплинути на подальше лікування, прогноз і профілактику патологій, у т. ч. спектр родини хворого (табл. 7).
|
Таблиця 7. Червоні прапорці сЦХДС (модифіковано згідно з Guey S. et al., 2021) [12] |
|
|
Червоні прапорці |
Додаткові зауваження |
|
Сімейний анамнез |
Необхідно перевірити сімейний анамнез інсульту, деменції або рухової недостатності. Сімейний анамнез може бути відсутнім у разі мутації de novo, автосомно-рецесивного успадкування, зниженої експресії або неповної пенетрантності |
|
Ранній початок інсульту |
Повторний інсульт із відносно значною кількістю маркерів візуалізації ЦХДС і відсутністю або дуже малою кількістю судинних факторів ризику |
|
Негативне дослідження на інші причини сЦХДС |
Значна кількість маркерів візуалізації ЦХДС, що контрастує з негативним результатом обстеження, і відносна нестача судинних факторів ризику. Однак наявність судинних факторів ризику не виключає діагнозу сЦХДС у пацієнтів з іншими червоними прапорцями відповідно до рекомендацій EAN |
|
Сугестивні позамозкові / системні особливості та клінічні й нейровізуалізаційні характеристики специфічного моногенного сЦХДС |
Ці клінічні прояви слід шукати не тільки в хворого, а й у членів його родини |
|
Кровна спорідненість |
Автосомно-рецесивне успадкування рідко зустрічається при сЦХДС, але кровне споріднення може свідчити про CARASIL |
Зазвичай МРТ може виявляти типові зміни, характерні для окремих патологій, які належать до сЦХДС (рис. 8).
![Рис. 8. а) МРТ у режимі T2 показує поширену лейкоенцефалопатію з вираженим ураженням передніх скроневих часток за CADASIL-синдрому (згідно з Locatelli M. et al., 2020) [24]; б) МРТ‑послідовність FLAIR показує типові інсультоподібні ураження (кортикальні та підкіркові гіперінтенсивності), що охоплюють двобічні тім’яні ділянки при MELAS-синдромі (згідно з Yi Shiau Ng et al., 2019) [33]](/multimedia/userfiles/images/2023/ZU_11_2023/ZU_11_2023_pic_8_st_16_17.jpg)
Рис. 8. а) МРТ у режимі T2 показує поширену лейкоенцефалопатію з вираженим ураженням передніх скроневих часток за CADASIL-синдрому (згідно з Locatelli M. et al., 2020) [24]; б) МРТ‑послідовність FLAIR показує типові інсультоподібні ураження (кортикальні та підкіркові гіперінтенсивності), що охоплюють двобічні тім’яні ділянки при MELAS-синдромі (згідно з Yi Shiau Ng et al., 2019) [33]
Далі буде.
