



19 липня, 2023
Фізіологічні антикоагулянти в організмі людини та механізм їхньої дії
Кров являє собою найважливішу інтегрувальну систему, яка забезпечує обмін метаболітами та інформацією між клітинами і тканинами. Протікаючи замкнутим контуром, вона виконує пластичну та захисну функцію, контактує з усіма органами, доставляючи кисень та поживні речовини. При ушкодженні кровоносної судини утворюються згустки, які зупиняють кровотечу. Система гемостазу потребує балансу між утворенням та розчиненням фібрину для запобігання вільному кровотоку в місцях ушкодження і для забезпечення перфузії крові через тканини. Занадто низьке згортання призводить до кровотечі, тоді як занадто високе може сприяти утворенню тромбів.
У місці ушкодження судин швидко відбуваються генерація тромбіну та утворення фібрину. З початком коагуляції інші речовини в крові (фізіологічні антикоагулянти) діють як гальмівні механізми, обмежуючи згортання в певній ділянці ушкодження, тим самим вони запобігають утворенню згустків (досить великих, щоб перешкоджати нормальному кровотоку).
Регуляція згортання проявляється на різних рівнях – або шляхом інгібування ферментів, або шляхом модуляції активності кофакторів. Найважливішими фізіологічними антикоагулянтами є антитромбін III (АТ), протеїн С (PC), протеїн S (PS), тромбомодулін (ТМ), кофактор гепарину II (КГII), інгібітор тканинного шляху згортання (TAFI), гепарин тощо [1, 2] (табл.).
|
Таблиця. Основні первинні фізіологічні антикоагулянти |
|
|
Антикоагулянт |
Механізм дії |
|
АТ III |
Прогресивно діючий інгібітор тромбіну, фактора Xa та меншою мірою інших факторів згортання. Плазмовий КГ |
|
PC |
Вітамін-К-залежний інгібітор факторів Va і VIIIa. Активується комплексом тромбін-ТМ |
|
PS |
Вітамін-К-залежний кофактор PC |
|
ТМ |
Рецепторний білок, що блокує тромбін, у комплексі з яким активує PC та PS |
|
КГII |
Утворює комплекс із гепарином. Особливо активний у плазмі, позбавленій АТ ІІІ |
|
TAFI |
Інгібітор комплексу ТФ-VIIa-Xa-Ca++ |
|
Гепарини |
Утворюють комплекси з АТІІІ, перетворюючи його на антикоагулянт |
|
Анексин V |
Один із мембранозв’язаних глікопротеїнів, що інгібує взаємодію ферментних факторів згортання крові між собою на фосфоліпідних мембранах клітин |
|
α2-макроглобулін, α1-антитрипсин |
Інгібітори лейкоцитарних протеаз. Слабкі інгібітори тромбіну, фактора XIa, калікреїну |
|
β2-глікопротеїн-1 |
Глікопротеїн, вбудований у фосфоліпідні мембрани клітин. Неспецифічний інгібітор факторів X та II |
|
Контактні інгібітори |
Порушують активацію факторів XII та XI |
|
Інгібітор комплементу-1 |
Слабкий інгібітор факторів XIa, XIIa, калікреїну, С1-компонента комплементу |
|
Інгібітори полімеризації фібрин-мономерів |
Гальмують утворення фібрину. Значення в регуляції гемостазу не встановлено |
Антитромбін III
На початку XX ст. завдяки дослідницьким роботам C. Contanjean, P. Morawitz і W. Howell виявлено, що такий фермент, як тромбін, поступово втрачає свою активність при його додаванні до дефібринованої плазми або сироватки [3-5]. Ґрунтуючись на цьому факті, було висловлено припущення, що специфічним інактиватором цього ферменту в плазмі за нормальних фізіологічних умов може бути АТ. У 1916 р. J. McLean виділив гепарин із печінки та серця і продемонстрував його потужну антикоагулянтну дію [6]. Докази щодо його інгібувальної дії на очищені прокоагулянти надали в 1939 р. К. Brinkhous і співавт., які показали, що гепарин ефективний як антикоагулянт тільки за наявності одного з компонентів плазми, що отримав назву КГ або АТ [7]. У роботах Waugh та співавт. і Monkhouse та співавт. [8, 9] відзначено тісний зв’язок між активністю АТ та гепарину в плазмі. Ці дослідники припустили, що гепарин прискорює у 50-100 разів швидкість, з якою АТ нейтралізує тромбін. У 1968 р. ця гіпотеза була остаточно підтверджена U. Abilgaard [10, 11].
Характеристика АТ ІІІ
АТ III синтезується в печінці та являє собою глікопротеїн із молекулярною масою 58 кДа, що складається з одного амінокислотного ланцюга (432 амінокислоти, на яких розташовані 4 N‑глікозильовані олігосахаридні ланцюги). Нормальна концентрація рівня АТ у плазмі крові людини становить ≈0,12 мг/мл, що відповідає молярній концентрації 2,3 мкмоль з періодом напіврозпаду ≈3 днів. АТ III являє собою інгібітор серинової протеази (SERine Protease INhibitors, SERPIN), який діє як псевдосубстрат для інгібування факторів IIa (тромбін), IXa, Xa, XIa, XIIa, а також калікреїну та плазміну за допомогою ковалентного зв’язування з активним центром серинової протеази. У плазмі циркулюють дві ізоформи АТ, а саме форми α і β, що не відрізняються одна від одної за ефектом інгібування тромбіну, проте вони різняться за своєю спорідненістю до гепарину. В нормальних умовах 85-95% циркулювального АТ є глікозильованими за 4 молекулами аспарагінової кислоти (належать до α-ізоформи). Інші 5-15% представлені β-ізоформою і мають більшу спорідненість до гепарину.
Механізм дії АТ ІІІ
Інактивація тромбіну АТ III відбувається шляхом утворення ковалентного зв’язку, внаслідок чого утворюється неактивний стехіометричний комплекс (1:1) між ними, що включає взаємодію активного серину тромбіну та реактивної ділянки аргініну на AT. Швидкість нейтралізації серинових протеаз під дією АТ протікає повільно без гепарину, але значно прискорюється за його наявності. Оскільки терапевтичний антитромботичний ефект гепарину опосередкований АТ, гепарин in vivo є неефективним за відсутності або майже відсутності АТ. У хворих зі спадковою недостатністю АТ після його введення він тимчасово замінює відсутній.
Мінімальною структурою, що забезпечує активацію АТ гепарином, є пентасахарид, що містить 4 специфічно розташовані сульфогрупи [12]. Активувальна дія такого пентасахариду зумовлена впливом на четвертинну структуру AT, а саме на положення петлі, що містить реактивний центр (рис. 1).
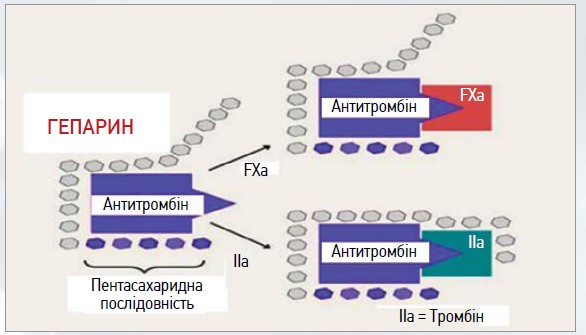
Рис. 1. Механізм антикоагулянтної дії АТ III
Гепарин із високою спорідненістю зв’язується з АТ через пентасахаридну послідовність, у результаті чого відбувається зміна його конформаційної структури, що призводить до прискорення взаємодії АТ із тромбіном (фактор IIa) або фактором Ха. Зміна рН та іонної сили значно впливає на результат цієї взаємодії, що свідчить про електростатистичну природу зв’язування [13]. Активація AT, яка забезпечується пентасахаридним фрагментом, що містить у певних положеннях сульфогрупи, є дуже важливим, але не єдиним механізмом, котрий визначає антикоагулянтну дію гепарину. Висока швидкість інгібування тромбіну (приблизно на порядок перевищує швидкість інгібування фактора Xa) обумовлена тим, що в цій реакції гепарин не тільки активує AT, а й завдяки зв’язуванню із тромбіном виконує роль матриці, забезпечуючи в такий спосіб ефективну взаємодію протеази з інгібітором.
Отже, для ефективного пригнічення протеаз АТ необхідний гепарин, який синтезується опасистими клітинами. У стінці судин та на люмінальній поверхні ендотелію наявна значна кількість глікозаміногліканів і глікопротеїнів, вуглеводні ланцюги яких мають гепаринові структури, що беруть участь в активації AT, а також нейтралізації тромбіну.
Дефіцит АТ III
Дефіцит АТ може бути набутим або спадковим.
Набутий дефіцит АТ виникає у результаті різних механізмів:
- посилення екскреції: ниркова недостатність, пов’язана з протеїнурією, нефротичний синдром, сильна крововтрата;
- зниження синтезу: синдром недостатності харчування, захворювання печінки, неонатальний період;
- збільшення споживання: ДВС‑синдром, венооклюзійна хвороба, оперативне втручання, прееклампсія, тяжка травма;
- застосування лікарських препаратів: оральні естрогени, L‑аспарагіназа, гепарин.
Причини набутого дефіциту АТ III виявити легше, ніж спадковий дефіцит [14, 15].
Спадковий дефіцит АТ ІІІ – це генетичне захворювання, що спричиняє згортання крові більше, ніж зазвичай. Належить до автосомно-домінантного розладу, за якого людина успадковує одну копію гена SERPINC1 на хромосомі 1q25.1, котрий кодує АТ. Цей стан призводить до підвищеного ризику венозних та артеріальних тромбозів із проявом клінічних симптомів зазвичай в юнацькому віці. Спадковий дефіцит АТ виникає тоді, коли людина отримує одну аномальну копію гена АТ III одного з батьків. Залежно від того, яка з функцій АТ порушена розрізняють кілька типів спадкового дефіциту: тип I характеризується тим, що приблизно однаково знижено функціональну активність АТ та його концентрацію. Це класичний та найпоширеніший тип дефіциту; тип ІІ – в пацієнтів концентрація АТ перебуває у межах норми, однак його властивості змінено.
Оскільки сам собою дефіцит АТ не обов’язково спричиняє розвиток тромбозів (носії можуть дожити до глибокої старості, не маючи тромботичних ускладнень), лікування слід рекомендувати за розвитку тромбозу або в разі наявності додаткових факторів ризику.
Показання щодо АТ III‑тестування:
- скринінг та діагностика дефіциту АТ;
- рецидивні тромбоемболічні явища;
- підозра на спадкову недостатність АТ;
- підозра на набутий дефіцит АТ;
- моніторинг при замісній терапії;
- відсутність відповіді терапією на гепарин.
Референсні значення АТ III: 83-128%.
Інтерпретація значень показників АТ ІІІ (% активності)
Підвищення значень: запальні процеси, гострий гепатит, холестаз, дефіцит вітаміну К, прийом антикоагулянтів, тяжкий панкреатит, рак підшлункової залози, менструація, лікування анаболічними препаратами.
Зниження значень (<70%): вроджений дефіцит, останній триместр вагітності, післяопераційний період, захворювання печінки (хронічний гепатит, цироз), гострий ДВС‑синдром, хронічна печінкова недостатність, тромбоемболія, сепсис, уведення гепарину, прийом пероральних контрацептивів.
Протеїн С
РС – вітамін-К‑залежний неактивний попередник серинової протеази, яка є ключовим компонентом у фізіологічно важливій природній антикоагулянтній системі.
Структура та функціональні аспекти
РС синтезується в печінці у вигляді довгої одноланцюгової послідовності, що містить 461 амінокислоту. До секреції він перетворюється на дволанцюговий поліпептид, що містить один легкий (155 амінокислот) і один важкий (262 амінокислоти) ланцюги, пов’язані дисульфідними зв’язками. Молекулярна маса зрілого білка становить ≈62 кДа. Циркулює у плазмі людини в концентрації 3-5 мкг/мл із періодом напіввиведення 6-8 год [16].
Активація PC
РС перетворюється на активну серинову протеазу, яка є фізіологічно функціональною. N‑кінцевий легкий ланцюг РС містить 9 γ-карбоксилованих залишків Glu (Gla-домен) і 2 домени, схожі на епідермальний фактор росту (EGF). Активація протеїну людини відбувається шляхом ферментативного видалення невеликого пептиду від важкого ланцюга [17]. У природних умовах активація опосередковується комплексом тромбін-ТМ [18, 19], а in vitro досягається з використанням тромбіну, трипсину або протеаз різних зміїних отрут [20-22].
Тромбін-ТМ
Активація РС лише тромбіном відбувається повільно та не має фізіологічної функції. Однак, коли тромбін зв’язується з інтегральним мембранним білком ТМ, наявним у судинах ендотелію, це призводить до збільшення швидкості у 20 000 разів, з якою тромбін активує РС [23]. Іони Ca2+ є важливим регулятором, які посилюють активацію РС за рахунок комплексу тромбін-ТМ та інгібують активацію тромбіну [24]. Тромбін, що надходить до мікроциркуляції, швидко зв’язується з ТМ і сприяє активації РС (рис. 2).
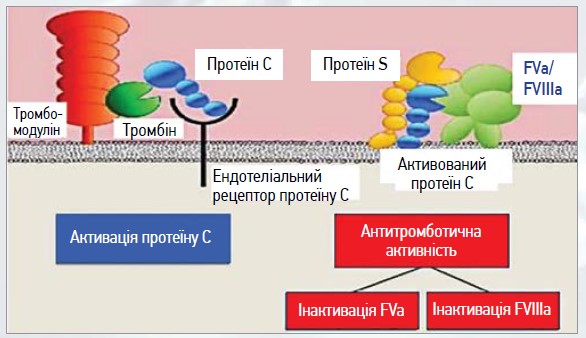
Рис. 2. Активація РС при зв’язуванні тромбіну із ТМ
Зміїні отрути
Відомо кілька зміїних отрут, які можуть активувати РС, зокрема з роду отруйних змій Agkistrodon (щитомордник). Три серинові протеази були виділені й охарактеризовані з отрут цього сімейства, що включає види A. bilineatus, A. h. halys та A. c. contortrix, ферменти яких є специфічними активаторами РС швидкої дії.
Функція активованого РС
Доведено антикоагулянтну функцію активованого РС (АРС) за рахунок подовження протромбінового часу та активованого часткового тромбопластинового часу. АРС розщеплює й інактивує 2 мембранозв’язані прокоагулянтні білки – фактор Va і фактор VIIIa, що зумовлює зниження утворення тромбіну (рис. 3).
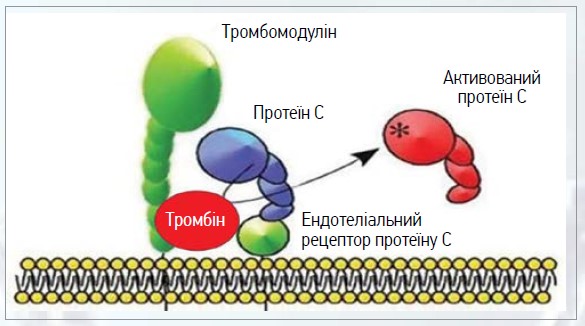
Рис. 3. Активований PС розщеплює й інактивує 2 мембранозв’язані прокоагулянтні білки – фактор Va і фактор VIIIa
Кофактори АРС
Молекулярні механізми, що беруть участь в антикоагулянтній системі РC, є дуже складними та досі не зовсім зрозумілими. Однак доведеними для прояву максимальної каталітичної ефективності АРС є кофактори, до яких належать РS і фактор V [25].
Протеїн S
PS – вітамін-К‑залежний білок плазми, який є також і кофактором APC; пов’язує регуляторний білок гострої фази системи комплементу C4bBP і опосередковує його приєднання до фосфоліпідів [26]. Приблизно 60% РS у плазмі зв’язується із C4bBP. Тільки вільна форма РS має функцію кофактора APC. РS посилює зв’язування APC із фосфоліпідами тромбоцитів та ендотеліальними клітинами.
Фактор V
Нещодавні дослідження показали можливу антикоагулянтну роль фактора V. АРС за наявності фактора V та РS ефективно розщеплює свої субстрати, тоді як APC окремо або разом із фактором V не є ефективним. Вважається, що РS і фактор V функціонують як синергетичні кофактори для APC у мультимолекулярному комплексі на поверхні мембрани.
Інгібітори АРС
APC інгібується відносно повільно, щонайменше трьома інгібіторами протеаз у плазмі, включаючи інгібітор РС (відомий як PAI‑3), інгібітор трипсину та α2-макроглобулін.
Дефіцит РС
Венозний тромбоз – серйозна медична проблема. Патогенез захворювання, найімовірніше, є багатофакторним і пов’язаний як із непрямими, так і зі спадковими чинниками ризику. Важливість виявлення патології, що лежить в основі генетичної причини, очевидна, оскільки до 40% пацієнтів із тромбозом мають позитивну сімейну історію.
Спадковий дефіцит протеїну
Актуальність оцінки рівня РС у плазмі пацієнтів стала очевидною з 1981 р., коли про перший випадок спадкового дефіциту РС, пов’язаного із тромботичною хворобою, повідомили Griffin і співавт. [27]. Подальші дослідження це підтвердили. Було показано, що в осіб з ізольованим дефіцитом РС ризик венозного тромбозу може збільшуватися в 6-9 разів [28, 29]. Спадковий дефіцит РС успадковується як автосомно-домінантна ознака. Гетерозиготи з дефіцитом РС мають активність лише на рівні від 30 до 70% від норми. В гомозигот із легким дефектом РC рівні знаходяться в межах 10-24%, а із сильним дефектом (компаунд-гетерозиготи) цей показник становить <1%.
Нормальні рівні РС
Нормальний рівень РС у дорослих складає від 70 до 130%.
Поширеність дефіциту РС
Поширеність дефіциту РС становить 2-5% у пацієнтів із тромбоемболією. Окремі групи хворих із тромбозом, що з’явився в молодому віці, зазвичай мають вищу поширеність (до 10-15%).
Клінічні особливості
Найчастішим клінічним проявом симптоматичного гетерозиготного дефіциту РС є тромбоз глибоких вен нижніх кінцівок. Перша тромботична подія зазвичай відбувається після статевого дозрівання. Серед груп пацієнтів (50-80%) виявляється у віці до 30-45 років. До важливих непрямих факторів ризику, пов’язаних з виникненням тромбозу, належать хірургічне втручання, іммобілізація, вагітність та прийом оральних контрацептивів. Однак ≈50% усіх перших тромботичних подій та 65% рецидивів виникають спонтанно без видимої причини. На відміну від гетерозиготного стану, який сам собою є відносно слабким фактором ризику тромбозу, пацієнти з гомозиготним дефіцитом РС зазвичай страждають від тяжких і фатальних тромбозів у ранньому віці. Клінічна картина проявляється у вигляді блискавичної пурпури, тромбозу головного мозку та ДВС‑синдрому. Гомозиготний дефіцит РС трапляється дуже рідко (1 на 500 000 народжень).
Стани та препарати, пов’язані зі зміною рівня РС
Знижені рівні: ДВС‑синдром, тромбоз глибоких вен, легенева емболія, захворювання печінки, післяопераційні пацієнти, інфекційні захворювання, рак, L‑аспарагіназна терапія, респіраторний дистрес-синдром, гемолітичний уремічний синдром, тромботична тромбоцитопенічна пурпура, неонатальний період. Підвищені рівні: діабет, нефротичний синдром, пізня вагітність, оральні контрацептиви.
Протеїн S
У 1977 р. при очищенні РС із бичачої плазми дослідники виявили новий білок, що містить γ-карбоксиглутамат (Gla), який вони назвали PS на честь Seattle (міста його винайдення) [30]. У наступні роки PS був визнаний білком із широким набором функцій.
Білок S – це вітамін-К‑залежний глікопротеїн плазми. PS секретується переважно ендотеліальними клітинами, мегакаріоцитами, гепатоцитами і клітинами Лейдіга [31]. Ці клітини спочатку синтезують PS у вигляді білка-попередника, що має 676 амінокислот [32], який після свого розщеплення та розщеплення пропептидів перетворюється на білок зі 635 амінокислотами з молекулярною масою 70 кДа, що складається з N‑кінцевого домену Gla, тромбонечутливої ділянки (TSR), чотирьох епідермальних ростових факторів (EGF‑подібних доменів) і глобуліну, що зв’язує статеві гормони (SHBG). PS циркулює у крові в концентрації 350 нмоль з періодом напіврозпаду 42 год. Близько 60% PS пов’язані з компонентом комплементу C4bBP, інші 40% є вільними [33]. Переважно біологічну активність виявляє лише вільна форма PS.
Антикоагулянтна функція PS
PS відомий як кофактор антикоагулянтної та профібринолітичної активності РС. Разом із РС і мембранним рецептором до РС становить т. зв. систему РС, яка забезпечує інгібування факторів згортання, як-от Va і VIIIa. Крім того, РS чинить і незалежну від АРС антикоагулянтну дію як кофактор TAFI, що інгібує комплекс тканинного фактора (VIIa-TF), отже, активацію фактора X [34].
Коферментна функція PS
Оскільки PS є кофактором АРС і TFPI, це виявляється в посиленні функції АРС у 3-10 разів, а TFPI – у 3-5 разів [35] (рис. 4).
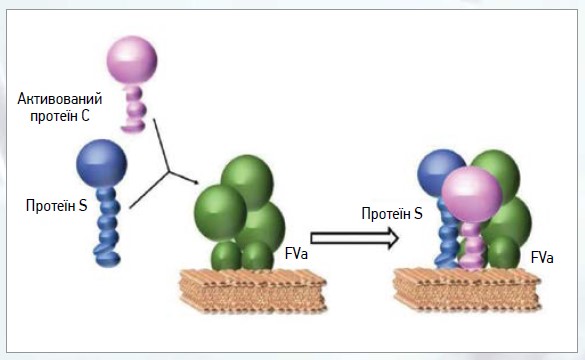
Рис. 4. Молекулярний механізм, що демонструє участь PS з активованим PC для інактивації фактора Va
Медична газета «Здоров’я України 21 сторіччя» № 11 (547), 2023 р
