


16 травня, 2022
Молнупіравір для перорального лікування амбулаторних пацієнтів із COVID-19
Пандемія коронавірусної хвороби 2019 (COVID‑19), зумовленої вірусом тяжкого гострого респіраторного синдрому (SARS-CoV‑2), спричинила майже 270 млн підтверджених випадків і понад 5,2 млн смертей в усьому світі. Госпіталізації потребує значна частина пацієнтів із COVID‑19, переважно люди старшого віку й особи з хронічними захворюваннями, як-от ожиріння, цукровий діабет і тяжка серцева патологія. На сьогодні схвалено низку вакцин, високоефективних у зниженні частоти госпіталізації та смертності; утім, охоплення вакцинацією залишається недостатнім. Отже, існує потреба в противірусній терапії, яка б знижувала ризик прогресування COVID‑19. Дослідження засвідчили важливість якомога ранішого початку лікування після появи симптомів, тому в ідеалі ця терапія має бути легкодоступною і такою, що може вільно застосовуватися самими пацієнтами.
Молнупіравір – низькомолекулярні рибонуклеозидні проліки N‑гідроксицитидину (N-ГЦ) – є активним проти SARS-CoV‑2 й інших РНК‑вмісних вірусів і має високу стійкість до розвитку резистентності. Після перорального застосування молнупіравіру N-ГЦ надходить у системний кровоток і фосфорилюється всередині клітин з утворенням N-ГЦ‑трифосфату, який вбудовується у вірусну РНК і порушує її реплікацію шляхом внесення помилок копіювання. Останні накопичуються у вірусному геномі та зрештою роблять вірус незаразним й унеможливлюють його реплікацію.
Молнупіравір оцінювали в низці досліджень I та ІІ фази. На підставі отриманих результатів обрали добову дозу 800 мг для подальшого вивчення, зокрема в дослідженні ІІІ фази MOVe-OUT, у якому взяли участь амбулаторні дорослі пацієнти групи ризику з появою симптомів COVID‑19 не раніше 5 днів до включення.
Методи
MOVe-OUT, подвійне сліпе рандомізоване плацебо-контрольоване дослідження ІІІ фази з оцінки безпеки й ефективності молнупіравіру в амбулаторних дорослих хворих на COVID‑19, розпочалося 6 травня 2021 р., коли перший пацієнт пройшов скринінг. На підставі позитивних результатів ефективності запланованого проміжного аналізу від 10 вересня 2021 р., коли отримали дані 29-денного спостереження 50% із 1550 пацієнтів (цільова вибірка), незалежний комітет із моніторингу рекомендував дочасно припинити набір пацієнтів.
У дослідження залучали дорослих пацієнтів із COVID-19 легкого та середнього ступеня тяжкості, які не потребували госпіталізації. Тяжкість хвороби встановлювали на основі визначень, адаптованих із настанов FDA та ВООЗ. Головними критеріями включення на момент рандомізації були інфекція SARS-CoV‑2, лабораторно підтверджена протягом останніх 5 днів, принаймні 1 ознака або симптом COVID‑19 та щонайменше 1 фактор ризику прогресування до важкої форми COVID‑19 (вік >60 років; активний рак; хронічна хвороба нирок; хронічна обструктивна хвороба легень; ожиріння, визначене як індекс маси тіла ≥30 кг/м2; серйозна патологія серця – серцева недостатність, ішемічна хвороба серця або кардіоміопатія; цукровий діабет). Основні критерії виключення: очікувана потреба в госпіталізації з приводу COVID‑19 в останні 48 год, діаліз або розрахована швидкість клубочкової фільтрації <30 мл/хв/1,73 м2, вагітність, небажання використовувати контрацептиви протягом прийому досліджуваного лікування та щонайменше 4 днів після його завершення, тяжка нейтропенія (абсолютний вміст нейтрофілів <0,5×109/л), вміст тромбоцитів <100×109/л), вакцинація проти SARS-CoV‑2. Стандартні лікувальні заходи, як-от антипіретики, протизапальні препарати, глюкокортикоїди та їхні комбінації, дозволялися. Натомість використання терапії, призначеної для лікування COVID‑19 (включно з моноклональними антитілами й ремдесивіром) заборонялося до 29-го дня спостереження.
Пацієнтів, котрі відповідали зазначеним критеріям, рандомізували 1:1 для отримання молнупіравіру (добова доза 800 мг; капсули 200 мг 4 рази на день) або плацебо перорально протягом 5 днів.
Вихідну наявність антитіл проти нуклеокапсидного білка SARS-CoV‑2 визначали централізовано за допомогою імуноаналізу Elecsys (Roche Diagnostics). Ознаки та симптоми COVID‑19 (відсутні, легкі, помірні або тяжкі) пацієнти оцінювали в щоденниках кожного дня – від рандомізації до 29-го дня спостереження. Для кількісного визначення РНК SARS-CoV‑2 у мазках із носоглотки застосовували полімеразну ланцюгову реакцію, генотипування виконували за допомогою секвенування наступного покоління.
Кінцеві точки
Первинною кінцевою точкою ефективності була частота госпіталізації з будь-яких причин (визначеної як ≥24 год невідкладної допомоги в лікарні або аналогічному закладі) чи смерті протягом 29 днів спостереження в модифікованій популяції ITT (від intention-to-treat), що складалася з усіх пацієнтів, котрі пройшли рандомізацію, отримали щонайменше 1 дозу молнупіравіру чи плацебо та не були госпіталізовані до прийому першої дози. Первинною кінцевою точкою безпеки визначили частоту небажаних подій (НП). Показники безпеки, включно із часткою пацієнтів із НП, оцінювали в популяції, що складалася з усіх учасників, котрі пройшли рандомізацію та отримали щонайменше 1 дозу молнупіравіру чи плацебо.
Вторинні кінцеві точки ефективності й безпеки ґрунтувалися на 11-бальній шкалі клінічного прогресування ВООЗ та суб’єктивних ознаках і симптомах COVID‑19 протягом 29 днів спостереження. Покращення або прогресування ознак і симптомів COVID‑19 визначали як будь-яке полегшення або погіршення відповідно, як порівняти з вихідною тяжкістю симптомів. Час до стійкого зникнення або полегшення симптомів визначали як кількість днів від рандомізації до першого з 3 послідовних днів відсутності або полегшення симптомів (без наступного погіршення до 29-го дня спостереження), час до прогресування ознак і симптомів – як кількість днів від рандомізації до першого з 2 послідовних днів погіршення. Пошукові кінцеві точки включали середні зміни вірусного навантаження SARS-CoV‑2 порівняно з вихідним показником.
Результати
Запланований проміжний аналіз охопив 775 учасників (54,1% усіх рандомізованих пацієнтів), яких залучили в 78 клінічних центрах 15 країн. Фінальна вибірка складалася загалом із 1433 пацієнтів, залучених у 107 клінічних центрах 20 країн. За виключенням статі (в групі молнупіравіру було дещо більше жінок), вихідні демографічні й клінічні характеристики були в цілому подібними в обох групах на момент як проміжного аналізу, так й аналізу всієї рандомізованої вибірки.
Учасники дослідження були значною мірою репрезентативними реальній популяції хворих на COVID‑19. Загалом 47,7% пацієнтів мали ознаки та симптоми ≤3 днів до рандомізації, в 44,5% хвороба мала помірно тяжкий характер. Найпоширенішими факторами ризику були ожиріння (73,7%), вік понад 60 років (17,2%) та цукровий діабет (15,9%). Вихідні антитіла до нуклеокапсидного білка SARS-CoV‑2, які свідчать про нещодавню або раніше перенесену інфекцію (не вакцинацію), були присутні в 19,8% учасників. Найчастішими варіантами SARS-CoV‑2 були B.1.617.2 (дельта; 58,1%), B.1.621 (мю; 20,5%) та Р.1 (гамма; 10,7%). Майже всі пацієнти (98,3%; 709 у групі молнупіравіру й 699 у групі плацебо) увійшли в модифіковану популяцію ITT. Серед учасників, котрі приймали молнупіравір або плацебо, більшість (95,2% та 94,7% відповідно) отримали щонайменше 9 доз.
Ефективність
На момент проміжного аналізу молнупіравір досяг попередньо встановленого критерію переваги; на 29-й день спостереження частка пацієнтів у модифікованій популяції ITT, котрі були госпіталізовані або померли, була значно меншою в групі молнупіравіру (7,3%), ніж у групі плацебо (14,1%), що відповідає абсолютній різниці між лікуванням -6,8% (95% довірчий інтервал – ДІ – від -11,3 до -2,4; р=0,001). У модифікованій ITT‑популяції всіх рандомізованих пацієнтів учасники, котрі отримували молнупіравір, також мали значно нижчий ризик госпіталізації або смерті протягом 29 днів: 6,8% проти 9,7% у групі плацебо (абсолютна різниця -3,0%; 95% ДІ від -5,9 до 0,1). Запланований аналіз, який оцінював лише госпіталізації та смерті, пов’язані з COVID‑19, показав, що таких випадків було 6,3% у групі молнупіравіру та 9,2% у групі плацебо (абсолютна різниця -2,8%; 95% ДІ від -5,7 до 0). Результати post hoc аналізу з поправкою на стать пацієнтів (єдиний вихідний фактор, потенційно незбалансований між групами) узгоджувалися з такими первинного аналізу: абсолютний ризик госпіталізації або смерті до 29-го дня був на 2,8% нижчим (95% ДІ від -5,7 до 0,1) для молнупіравіру порівняно з плацебо. Первинним результатам також відповідали результати аналізу часу до події; частота госпіталізації або смерті протягом 29 днів була приблизно на 31% нижчою для молнупіравіру порівняно з плацебо (відносний ризик 0,69; 95% ДІ 0,48-1,01) (рис. 1). У групі молнупіравіру зафіксували лише 1 випадок смерті (29-денна загальна летальність 0,1%), натомість у групі плацебо померли 9 хворих (29-денна загальна летальність 1,3%), що відповідає на 89% (95% ДІ 14-99) нижчому ризику смерті для молнупіравіру проти плацебо. Усі 10 учасників були госпіталізовані перед тим, як померти, і всі летальні випадки були розцінені дослідниками як зумовлені COVID‑19.
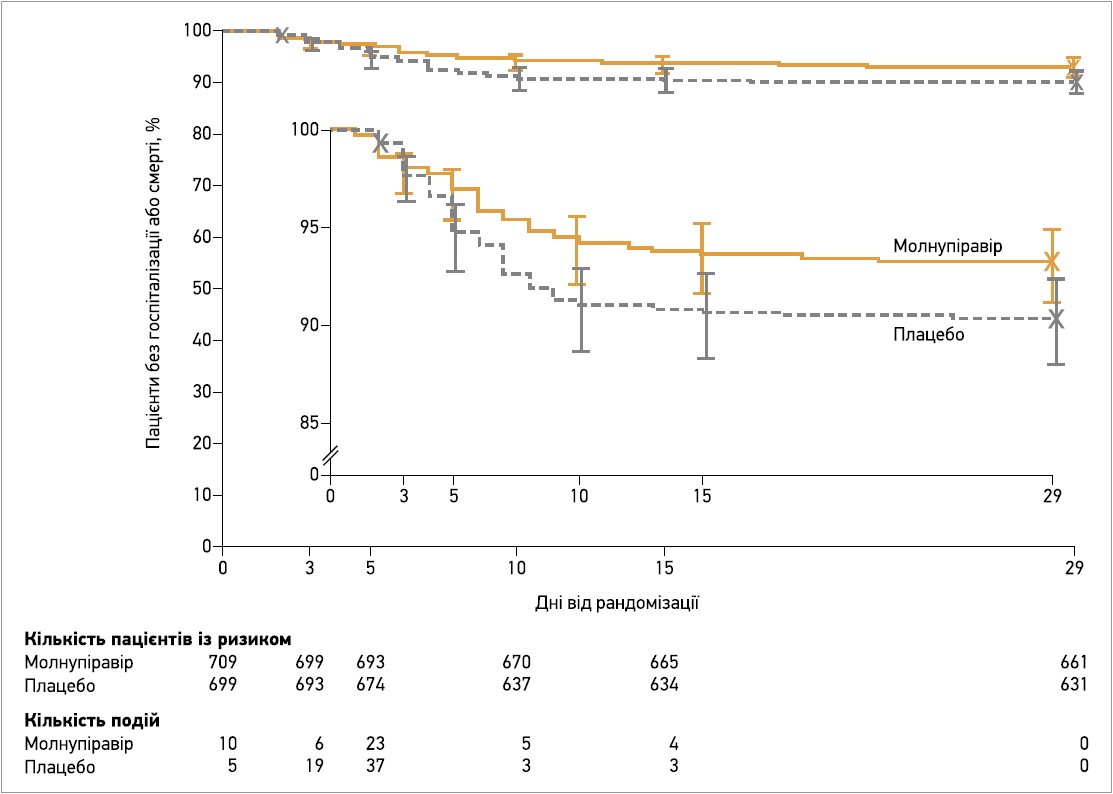
Рис. 1. Аналіз часу до події (госпіталізації або смерті) до 29-го дня в модифікованій популяції ITT
У більшості попередньо визначених підгруп частка пацієнтів, котрі були госпіталізовані або померли, була нижчою для молнупіравіру порівняно з плацебо, проте відповідні довірчі інтервали свідчать про значну невпевненість щодо ступеня цих ефектів (рис. 2). Імовірна різниця на користь плацебо спостерігалася лише в пацієнтів, які на момент включення мали антитіла до нуклеокапсидного білка SARS-CoV‑2, низьке вірусне навантаження або страждали на діабет, а також у хворих азійської, негроїдної, корінної американської або змішаної негроїдно-європеоїдно-американської раси; утім, усі відповідні 95% довірчі інтервали включали нуль і були доволі широкими, зокрема через відносно невелику кількість пацієнтів у деяких із цих підгруп.
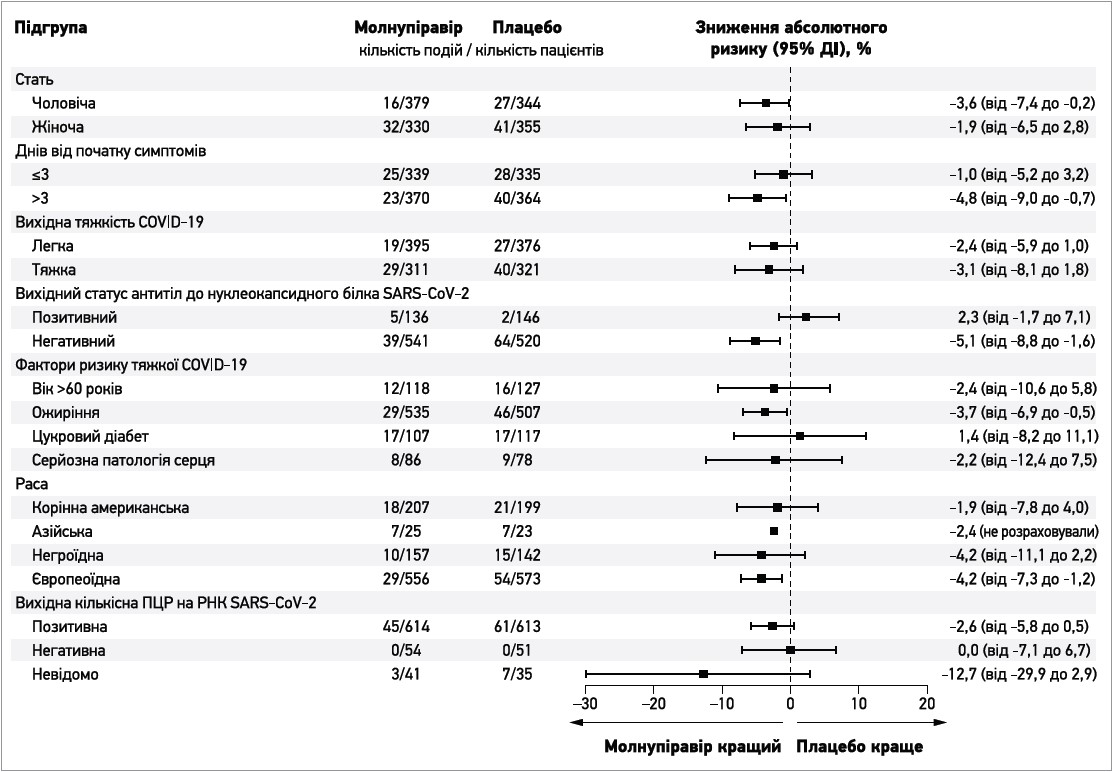
Рис. 2. Частота госпіталізації або смерті на 29-й день у модифікованій популяції ITT відповідно до підгруп
На підставі шкали прогресування ВООЗ у групі молнупіравіру більше пацієнтів порівняно з групою плацебо відмітили покращення на 5-й день, водночас найбільшу різницю на користь молнупіравіру спостерігали на 10-й і 15-й день. Для більшості симптомів та ознак COVID‑19 стійке зникнення або полегшення було більш імовірним, а прогресування – менш імовірним у групі молнупіравіру порівняно з групою плацебо.
На час проведення аналізу всіх рандомізованих пацієнтів 1093 з 1408 учасників (77,6%) у модифікованій популяції ITT мали кількісно визначену РНК, підтверджену в мазках із носоглотки на момент включення; зразки 964 пацієнтів (88,2%) були протестовані на 5-й день. З огляду на пошуковий характер вірологічної кінцевої точки тестування продовжується. Наявні на сьогодні дані свідчать, що лікування молнупіравіром асоціювалося з більшим зниженням вірусного навантаження порівняно з плацебо на 3-й, 5-й та 10-й день спостереження. Результати для інших часових проміжків були однаковими у двох групах.
Безпека
Частка пацієнтів із щонайменше 1 НП була подібною у двох групах (30,4% у групі молнупіравіру та 33,0% у групі плацебо), так само як і частка пацієнтів із НП, розцінених дослідниками як такі, що пов’язані з призначеним лікуванням (8,0% vs 8,3% відповідно). Випадки смерті внаслідок НП, жодна з яких, за оцінкою дослідника, не була пов’язана з лікуванням, траплялися рідше в групі молнупіравіру, ніж у групі плацебо (табл.).
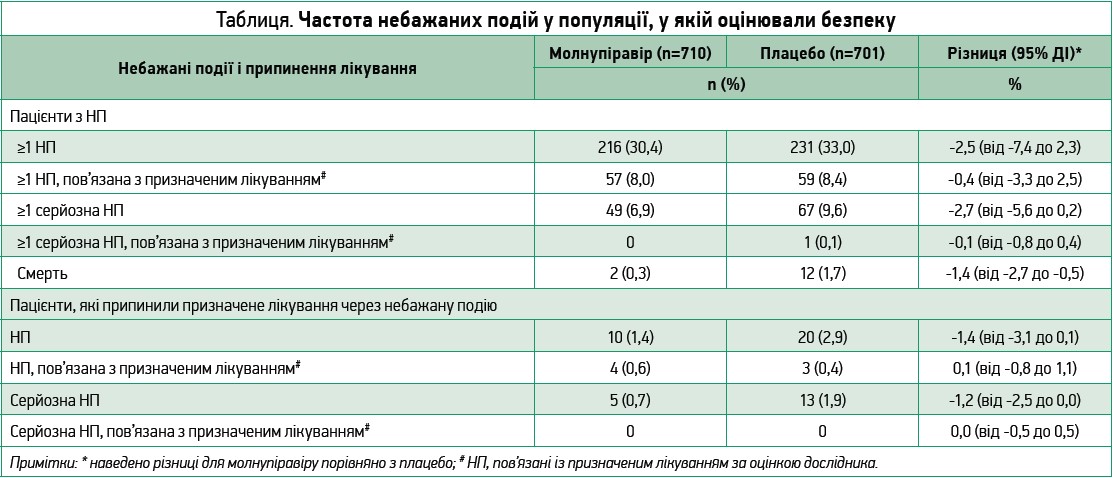
Найчастішими НП (такими, що спостерігалися у ≥2% пацієнтів однієї чи другої групи) були ковідна пневмонія (6,3% у групі молнупіравіру vs 9,6% у групі плацебо), діарея (2,3% vs 3,0%) та бактеріальна пневмонія (2,0% vs 3,0%); погіршення COVID‑19 як НП відзначили в 7,9% vs 9,8% пацієнтів відповідно.
Найчастішими НП (такими, що спостерігалися в ≥1% пацієнтів однієї чи другої групи), розціненими як пов’язані з досліджуваним лікуванням, були діарея (1,7% у групі молнупіравіру vs 2,1% у групі плацебо), нудота (1,4% vs 0,7%) та запаморочення (1,0% vs 0,7%). Зниження рівня тромбоцитів <50×109/л зафіксовано в 1 учасника кожної групи; в групі молнупіравіру низький рівень тромбоцитів спостерігався на 12-й день і не був пов’язаний із лікуванням.
Обговорення
Результати дослідження ІІІ фази MOVe-OUT за участю амбулаторних дорослих хворих на COVID‑19 із факторами ризику свідчать, що лікування молнупіравіром, розпочате в межах 5 днів від появи симптомів, знижує ризик госпіталізації з будь-яких причин або смерті протягом 29 днів. Популяція дослідження була репрезентативною реальній популяції хворих з одним або більшою кількістю відомих факторів ризику тяжкого перебігу COVID‑19.
В опублікованих клінічних дослідженнях, у яких вивчали моноклональні антитіла в подібних популяціях амбулаторних хворих на COVID‑19, частота госпіталізації або смерті в групах плацебо варіювала від 3% до 7%. Натомість в обговорюваному дослідженні цей показник становив 14% у проміжному аналізі та 10% в аналізі всіх рандомізованих пацієнтів; це свідчить, що учасники зазначеного дослідження мали вищий ризик прогресування хвороби.
Абсолютний ризик госпіталізації або смерті протягом 29 днів у групі молнупіравіру був на 6,8% нижчим порівняно з групою плацебо в проміжному аналізі та на 3,0% нижчим в аналізі всіх рандомізованих пацієнтів. Таке покращення є потенційно значимим для пацієнтів, систем охорони здоров’я та громадського здоров’я загалом. Ефективність молнупіравіру була послідовною в багатьох підгрупах, включно з пацієнтами, інфікованими варіантами SARS-CoV‑2 дельта, гамма та мю.
Вторинні кінцеві точки, як-от зміни оцінки за шкалою клінічного прогресування ВООЗ та суб’єктивних симптомів COVID‑19, також свідчать про клінічну користь від застосування молнупіравіру порівняно з плацебо.
Як і в попередніх дослідженнях, не спостерігалося жодних побоювань стосовно безпеки молнупіравіру, зокрема щодо клінічно значимих відхилень лабораторних показників. Оскільки вагітність була критерієм виключення з участі в дослідженні, потенційний вплив молнупіравіру на розвиток плода невідомий.
Багато пацієнтів із COVID‑19 одужують від гострої інфекції з мінімальним медичним втручанням або без такого. Утім, клінічне прогресування до тяжкої хвороби має суттєвий вплив на пацієнтів і системи охорони здоров’я, підвищуючи потребу в механічній вентиляції легень і ризик смерті та потенційно перевантажуючи локальні й регіональні потужності лікарень під час спалахів COVID‑19. Відтак, зменшення зумовлених COVID‑19 госпіталізацій, а також потенційне зниження передачі вірусу завдяки швидшому одужанню є критично важливими.
Вакцинація залишається найважливішим наявним медичним втручанням для зниження ризиків госпіталізації та смерті від COVID‑19, однак раннє, розпочате невдовзі після появи перших симптомів, лікування також є ефективним. Для лікування амбулаторних пацієнтів із COVID‑19 схвалені моноклональні антитіла бамланівімаб-етезевімаб, казиривімаб-імдевімаб та сотровімаб. Проте ці препарати призначаються шляхом інфузії або ін’єкції в умовах лікарні, тому пероральні засоби, як-от молнупіравір, який може застосовуватися пацієнтом у домашніх умовах незабаром після встановлення діагнозу, є більш практичним і дружнім до пацієнта лікуванням, якщо немає негайної потреби в госпіталізації. Отже, молнупіравір є цінним доповненням до наявного терапевтичного арсеналу при COVID‑19.
Ще однією перевагою молнупіравіру порівняно з моноклональними антитілами, спрямованими проти S‑білка SARS-CoV‑2, є його ефективність щодо різних варіантів вірусу. Механізм дії препарату не залежить від мутацій S‑білка, які можуть впливати на дієвість моноклональних антитіл.
У дослідження MOVe-OUT залучали тільки невакцинованих проти COVID‑19 пацієнтів, щоб, по-перше, зосередитися на хворих, котрі найімовірніше потребують противірусної терапії, а по-друге, для швидшої оцінки терапевтичної ефективності молнупіравіру. Відтак, потенційна користь препарату в лікуванні випадків COVID‑19, що розвинулися попри вакцинацію, не оцінювалася; показники ефективності в серопозитивних на момент включення пацієнтів не можна використовувати для остаточних висновків, оскільки тест на антитіла до нуклеокапсидного білка SARS-CoV‑2 відображає імунну відповідь на попередню або поточну інфекцію та не виявляє наявність створених вакциною нейтралізуючих анти-S антитіл.
Таким чином, дослідження MOVe-OUT продемонструвало, що в амбулаторних невакцинованих дорослих хворих на COVID-19 із факторами ризику пероральне лікування молнупіравіром, розпочате в межах 5 днів від появи симптомів, є ефективним і безпечним.
Стаття друкується у скороченні.
Список літератури знаходиться в редакції.
Bernal A.J., Gomes da Silva M.M., Musungaie D.B., et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N Engl J Med. 2022 Feb 10; 386 (6): 509-520.
Переклав з англ. Олексій Терещенко
