


1 жовтня, 2017
Периферична міопатія як терапевтична «мішень» при хронічній серцевій недостатності
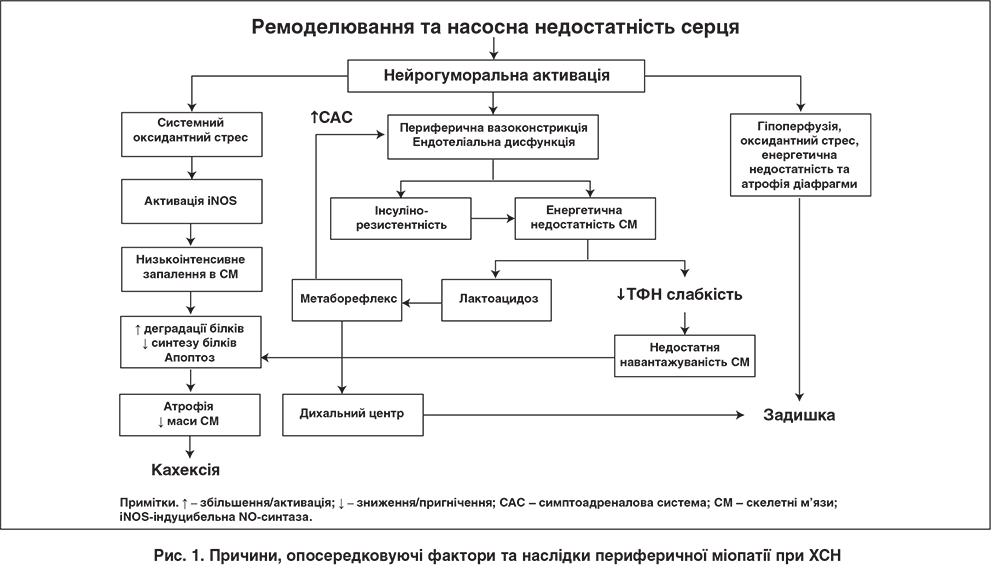
Процеси, що відбуваються в скелетних і дихальних м’язах при хронічній серцевій недостатності (ХСН), відіграють надзвичайно важливу роль у патофізіології цього синдрому, впливаючи не тільки на формування відповідної клінічної симптоматики, а й на його прогресування та клінічний прогноз.
Основною структурно-функціональною одиницею міоцитів поперечно-смугастих м’язів є міофібрили, які забезпечують акт їх скорочення, використовуючи для цього аденозинтрифосфорну кислоту (АТФ), що утворюється, як і в міокарді, з двох джерел – вуглеводів (переважно глюкози) та жирних кислот [1].
У дорослої людини поперечно-смугасті м’язи представлені трьома основними типами міофібрил – І, ІІа та ІІб, структурно-функціональна відмінність яких визначається ізоформами так званих тяжких ланцюгів локалізованого в них скорочувального білка – міозину [2]. Порівняно з І типом міофібрил волокна ІІб типу утворюють АТФ анаеробним (без участі кисню) шляхом ензиматичного перетворення глюкози, використовують продуковану енергію швидко, швидко ж скорочуються та «задіяні» за фізіологічних умов при виконанні коротких високоінтенсивних м’язових зусиль, для яких характерне швидке настання втоми. Останнє визначається не тільки швидким використанням АТФ, а й значно меншою продуктивністю гліколітичного (анаеробного) шляху її утворення порівняно з окислювальним шляхом (аеробним). Тип ІІа міофібрил характеризується як проміжний, оскільки міофібрили цього типу використовують одночасно обидва, окислювальний та анаеробний, шляхи утворення енергії й здатні забезпечувати як швидке (більшою мірою), так і повільне скорочення [2]. У нормі в скелетних м’язах (СМ) домінують міофібрили І типу [3], а ІІа тип переважає в діафрагмі [4].
При ХСН унаслідок зниження насосної спроможності серця обмежується можливість зростання серцевого викиду під час виконання фізичного навантаження, що теоретично є базовою передумовою для невідповідності перфузії СМ їхнім функціональним потребам. Утім, цей механізм є актуальним лише для пацієнтів із незворотною тяжкою ХСН (стадія D, фінальна, за класифікацією Американської колегії кардіологів (ACC) / Американської асоціації серця (АНА) – АСС/АНА) [5]. За сучасними уявленнями, основною причиною гіпоперфузії СМ при ХСН є істотне обмеження вазодилататорного резерву периферичних артерій на тлі дезадаптивних судинних ефектів, спричинюваних нейрогуморальною активацією (індуковані ангіотензином ІІ та норадреналіном вазоконстрикція й ремоделювання артерій і особливо пригнічення вазодилататорної функції ендотелію) [6-9]. Як наслідок, якщо під час виконання пікового фізичного навантаження кровоток у нижніх кінцівках у здорових осіб може збільшуватися майже у 20 разів, то в пацієнтів із ХСН – лише у 2-3 рази [6, 10]. Необхідно додати, що енергетичній недостатності СМ сприяє їхня інсулінорезистентність [11], яка спостерігається майже в половини хворих на ХСН за відсутності цукрового діабету [12].
Наслідком гіпоперфузії (і в багатьох випадках – інсулінорезистентності) СМ при ХСН є їхня хронічна енергетична недостатність у вигляді виснаження в міоцитах, за даними магнітно-резонансної (МР) спектроскопії, пулу креатинфосфату та зростання внутрішньоклітинної концентрації лактату внаслідок активації анаеробного шляху утворення АТФ [13, 14]. Паралельно відбувається пригнічення активності ензимів, які забезпечують аеробний (окислювальний) шлях енергетичного метаболізму [15, 16], що поєднане з відповідною трансформацією структурно-функціонального фенотипу СМ у вигляді зменшення в них кількості мітохондрій, загальної площини мітохондріальних крист [17] і збільшення порівняно з нормою кількості гліколітичних міофібрил ІІ типу з одночасним зменшенням кількості міофібрил І типу [18, 19]. Зростання внаслідок накопичення лактату концентрації водневих іонів спричиняє дисфункцію кальцієвого насоса (Са2+-АТФаза саркоплазматичного ретикулуму), який забезпечує узгодження між процесами розслаблення та скорочення міофібрил [20].
Клінічним відображенням зазначених змін є зниження сили й витривалості периферичних м’язів у пацієнтів із ХСН, яке корелює з функціональним класом (ФК) за NYHA, зумовлює скарги на слабкість і спричиняє зменшення толерантності до фізичного навантаження (ТФН) за даними функціональних тестів [21-23].
Активація при навантаженнях анаеробного метаболізму в СМ, яка супроводжується швидким настанням лактоацидозу, за механізмом метаборефлексу (синонім – ергорефлекс) спричиняє підвищення центрального симпатичного тонусу, внаслідок чого можливе збільшення адренергічних вазоконстрикторних стимулів із відповідним зменшенням кровотоку в нижніх кінцівках [24]. У такий спосіб метаболічні зміни в СМ, пов’язані з їхньою енергетичною недостатністю, замикають відповідне «хибне коло» нейрогуморальних і метаболічних чинників при ХСН-індукованій периферичній міопатії. З другого боку, зазначена активація анаеробної складової метаболізму СМ із виникненням лактоацидозу навіть за помірного навантаження через той самий метаборефлекторний механізм стимулює дихальний центр, унаслідок чого збільшується частота дихальних рухів і глибина дихання [25]. За допомогою МР-спектроскопії у пацієнтів із ХСН продемонстровано обернений кореляційний зв’язок між рівнем локального рН у СМ та об’ємом вентиляції легень за 1 хв під час виконання фізичного навантаження [26]. Отже, дизметаболізм СМ під час виконання фізичного навантаження є одним із чинників задишки при ХСН [25].
Іншим кардинальним напрямом негативного впливу нейрогуморальної активації на процеси в СМ при ХСН є стимулювання в них ангіотензином ІІ, а також певною мірою норадреналіном, оксидантного (вільнорадикального стресу) [5, 27]. Утворювані в надмірній кількості вільні радикали стимулюють локальну експресію в міоцитах індуцибельної NO-синтази (іNOS) – ензиму, за допомогою якого утворюється оксид азоту в патогенних (цитотоксичних) концентраціях [28]. Одним із важливих механізмів негативного впливу продукованого за допомогою іNOS оксиду азоту на скелетно-м’язові міоцити є пригнічення активності мітохондріальної креатинкінази – ферменту, що відповідає за транспорт синтезованої в мітохондріях енергії у вигляді фосфокреатину в цитозоль клітин [29], що, відповідно, поглиблює їхній енергодефіцит.
Через підвищення активності іNOS у макрофагах оксидантний стрес виступає тригером іншого важливого механізму ураження СМ при ХСН – системного запалення низької інтенсивності у вигляді зростання продукції макрофагами прозапальних цитокінів – фактора некрозу пухлини-альфа, інтерлейкінів 1 та 6 [30]. Наслідками згаданого низькоінтенсивного запалення в СМ є інтенсифікація процесів деградації скелетно-м’язових протеїнів поряд із пригніченням синтезу останніх [5]. Окрім запальної реакції, ще однією причиною змін при ХСН білкового обміну катаболічного спрямування в СМ є недостатня навантажуваність (англ. disuse) останніх унаслідок зниження фізичної активності пацієнтів [31]. У результаті поступово розвивається атрофія та спостерігається зниження маси СМ (саркопенія), що фундаментально притаманне синдрому ХСН і маніфестується в частини пацієнтів типовими ознаками кахексії [32].
Діафрагма, котра є основним дихальним м’язом, не лишається «осторонь» патологічних процесів, які відбуваються в поперечно-смугастій мускулатурі при ХСН. Особливостями діафрагми порівняно з м’язами кінцівок є її вельми потужна васкуляризація [33] й домінування в нормі проміжного (ІІа) типу міофібрил [4], що потенційно має забезпечувати їй більші адаптивні можливості в умовах ХСН. Більше того, при ХСН у діафрагмі відбуваються зміни з боку міофібрилярного апарату – протилежні тим, що спостерігаються при ХСН у СМ, а саме – збільшення кількості міофібрил І (аеробного) та зменшення кількості міофібрил ІІб (анаеробного) типів із відповідним зростанням активності окислювальних ензимів і пригніченням активності ензимів гліколітичних [34, 35]. Утім, попри наведені структурно-функціональні особливості та адаптаційні реакції діафрагми при ХСН, у ній урешті-решт розвиваються зміни, подібні до тих, що відбуваються в периферичних м’язах. Це, ймовірно, пов’язано зі значним, перманентним за своїм характером навантаженням, яке на неї припадає. У діафрагмі таких пацієнтів поступово розвивається енергетична недостатність, знижується функція мітохондрій, у тому числі зменшується експресія згаданої вище мітохондріальної креатинкінази, спостерігаються атрофічні зміни [5, 36]. У результаті знижуються як сила скорочень, так і витривалість дихальної мускулатури [37, 38], що вважається передумовою до компенсаторного збільшення частоти дихальних рухів [39]. Продемонстрована достовірна лінійна кореляція між показниками функції дихальної мускулатури та вираженістю задишки за шкалою Борга під час виконання стандартного (25 Вт) фізичного навантаження [39].
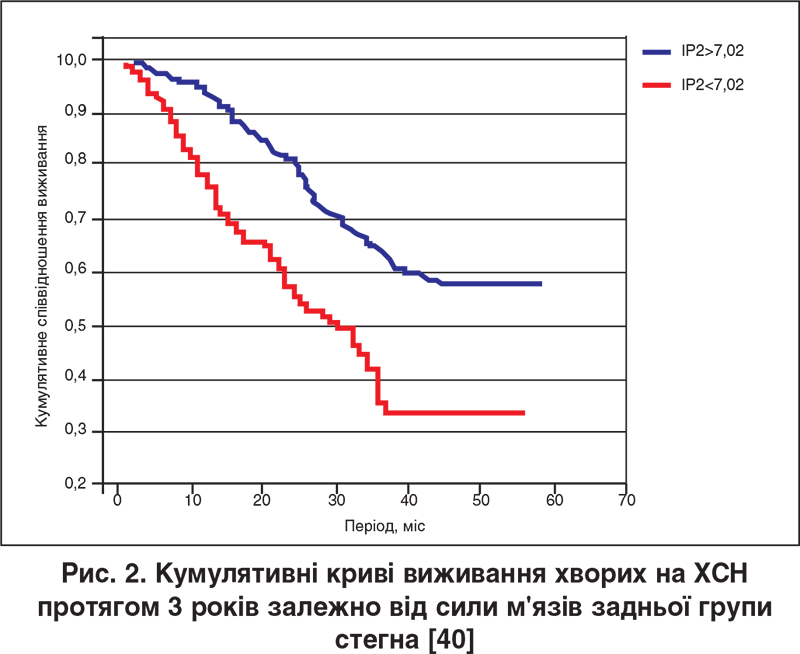
Усе викладене вище свідчить про те, що периферична міопатія при ХСН являє собою складний патофізіологічний феномен, який реалізується в таких його головних клінічних проявах, як зниження ТФН та поступово прогресуюча втрата маси СМ. Клінічна значущість цього феномена підкріплюється даними щодо прямого зв’язку між ступенем дисфункції м’язів нижніх кінцівок і наступною смертністю пацієнтів із ХСН при тривалому спостереженні (рис. 2) [40, 41].
Наразі цілісної стратегії, спрямованої на уповільнення прогресування периферичної міопатії при ХСН, не існує. Втім, на сьогодні накопичено певні дані щодо ефективності окремих фармакологічних і немедикаментозних підходів до покращення ТФН і зменшення інших проявів периферичної міопатії при ХСН. З-поміж стандартних рекомендованих при ХСН фармакологічних засобів позитивний терапевтичний вплив на механізми та прояви периферичної міопатії справляють блокатори ренін-ангіотензинової системи (РАС). Тривала – впродовж 6 міс – терапія пацієнтів із ХСН як інгібітором ангіотензинперетворювального ферменту (АПФ), так і сартаном супроводжувалася достовірним зростанням ТФН і асоціювалася (за даними повторних біопсій СМ) нормалізацією в останніх співвідношення типів міофібрил у вигляді достовірного збільшення кількості волокон I (аеробного) типу з відповідним зменшенням кількості гліколітичних волокон ІІб типу; зазначені зміни корелювали зі ступенем приросту пікового фізичного навантаження [42]. В іншому дослідженні зростання м’язової витривалості пацієнтів із ХСН на тлі терапії інгібітором АПФ корелювало з ефектом збільшення кількості мітохондрій у міофібрилах на тлі зазначеного лікування [43]. Як потенційні механізми вищезгаданого впливу блокаторів РАС на стан СМ при ХСН обговорюють їх здатність збільшувати кровоток у СМ завдяки вазодилататорному ефекту [44], а також пригнічувати гуморальну ланку імунозапальної відповіді [45].
За даними відповідного метааналізу, бета-блокатори при ХСН загалом не впливають у таких пацієнтів на симптоматику з боку СМ (слабкість, втомлюваність) [46]. З другого боку, відомо, що селективні бета-блокатори не підвищують ТФН у таких пацієнтів [47, 48]. Утім, досвід застосування карведилолу – бета-блокатора з вазодилататорними властивостями й антиоксидантною дією [49] – засвідчує його здатність збільшувати дистанцію в тесті з 6-хвилинною ходьбою, максимальну довільну силу м’язів нижніх кінцівок [50], підвищувати «суху» м’язову масу [51], що поєднується зі збільшенням на тлі його застосування ендотелійзалежної судинорозширювальної відповіді та швидкості кровотоку в тильній артерії стопи [52].
Аеробні фізичні тренування в пацієнтів із ХСН та зниженою фракцією викиду лівого шлуночка (ФВ ЛШ) у вигляді контрольованого регулярного виконання навантажень невисокої інтенсивності покращують ендотеліальну функцію, функцію ЛШ, підвищують ТФН і поліпшують якість життя [53, 54]. Окрім того, згідно з результатами метааналізу вони знижують ризик госпіталізацій із будь-яких причин, демонструючи тенденцію до зменшення смертності серед пацієнтів із тривалістю спостереження понад 1 рік [55]. Аеробні тренування з різними типами вправ супроводжуються підвищенням параметрів переносимості фізичного навантаження, а також, за даними виконаних у динаміці біопсій СМ, збільшенням кількості міофібрил І типу та щільності мітохондрій, чому відповідає покращення біохімічних індикаторів окислювального метаболізму [56-58]. Більше того, продемонстровано, що фізичні тренування тварин з експериментально змодельованою ХСН та істотно підвищеною експресією в СМ міостатину (м’язового протеїну – провідника катаболічних процесів) нормалізують експресію останнього до рівня, який спостерігався до процедури моделювання ХСН [59]. Широкому впровадженню тренувальних прог-
рам при ХСН можуть перешкоджати такі чинники, як брак відповідної медичної інфраструктури й труднощі під час формування прихильності багатьох пацієнтів до участі в таких програмах [5].
Зважаючи на ключову роль енергетичної недостатності скелетних і дихальних м’язів при ХСН, логічним вбачаємо випробування в таких пацієнтів поряд з іншими підходами терапевтичної стратегії, безпосередньо спрямованої на корекцію зазначених порушень енергетичного метаболізму. В цьому аспекті нас зацікавила можливість застосування екзогенного фосфокреатину з метою покращення клінічної симптоматики та підвищення переносимості фізичних навантажень у пацієнтів із тяжкою (III-IV ФК за NYHA) ХСН і низькою (<35%) ФВ ЛШ, що отримують оптимізоване стандартне лікування нейрогуморальними антагоністами та діуретиками.
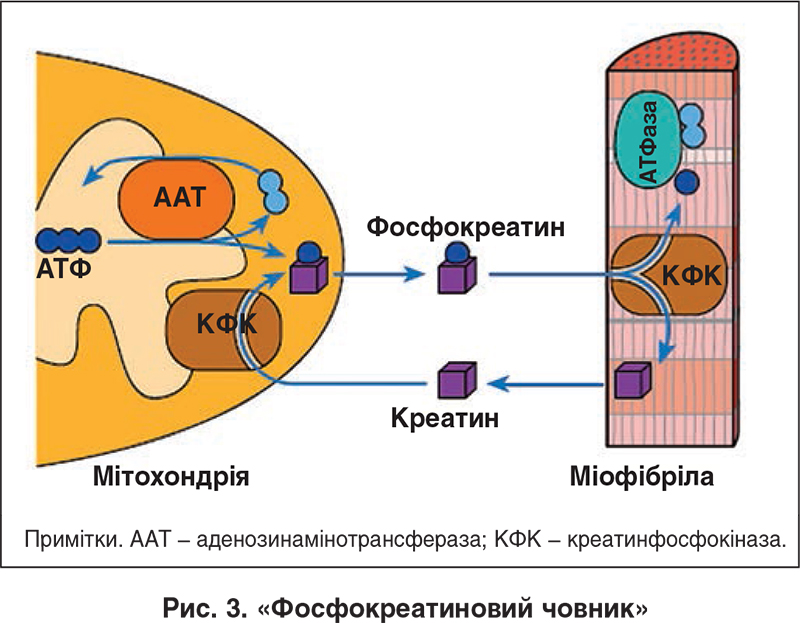
Процес переміщення АТФ, спродукованої в процесі окислювальних реакцій у мітохондріях, крізь мембрану останньої до місць її утилізації в саркоплазмі дістав назву «фосфокреатиновий човник». Ключовим компонентом функціонування останнього є згадувана вище мітохондріальна креатинкіназа (синонім – креатинфосфокіназа), здатна передавати енергію кінцевих фосфатних зв’язків від АТФ до креатину з утворенням фосфокреатину та аденозиндифосфату (АДФ). Швидко дифундуючи з митохондрій у саркоплазму, фосфокреатин транспортує енергію до місць її споживання (міофібрили, іонні насоси), де локальна креатинкіназа здійснює зворотне перенесення енергії кінцевого фосфатного зв’язку від фосфокреатину до АДФ, забезпечуючи в такий спосіб ресинтез АТФ із фосфокреатину. При цьому креатин, що звільнився, транспортується у зворотному напрямку в мітохондрію [60, 61] (рис. 3).
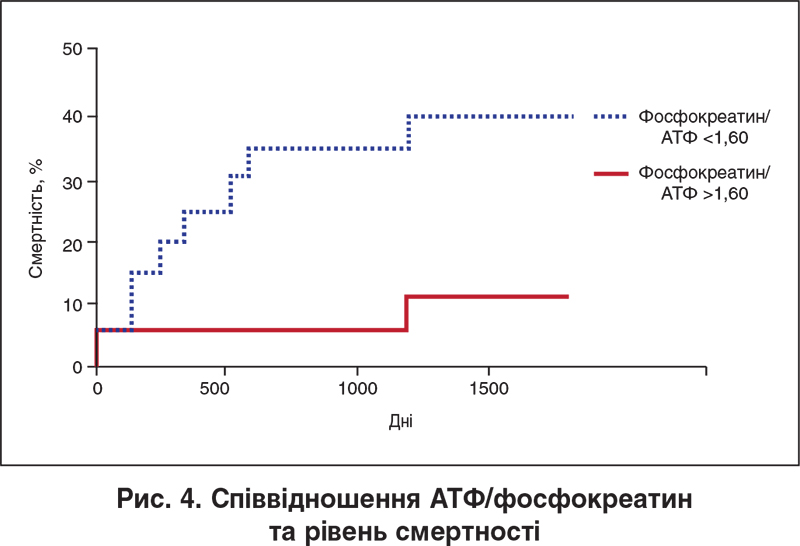
Окрім зменшення синтезу АТФ у клітинах міокарда та СМ, при СН спостерігається порушення згаданого вище механізму внутрішньоклітинного транспортування енергії, в основі якого лежить пригнічення активності згадуваної вище мітохондріальної креатинкінази [62]. Внутрішньоклітинне співвідношення фосфокреатин/АТФ (у нормі становить 1,7-2,1) розглядається як інтегральний маркер стану енергетичного обміну м’язових клітин, оскільки відображає адекватність як продукції АТФ, так і транспортування енергії в саркоплазму [60]. При ХСН спостерігається зниження цього співвідношення в міокарді пропорційно до її клінічної тяжкості (ФК за NYHA) [63], причому його зниження корелює із загальною смертністю та смертністю від серцево-судинних причин серед таких пацієнтів [64] (рис. 4). Зниження співвідношення АТФ/фосфокреатин спостерігається, за даними МР-спектроскопії, також і в СМ пацієнтів із ХСН, причому часткове відновлення зниженого вмісту фосфокреатину в СМ фіксувалося в них після фізичних тренувань упродовж 8 тиж [65].
Ідея застосування екзогенного фосфокреатину з метою швидкого відновлення його внутрішньоклітинного пулу знайшла своє втілення в низці експериментальних і клінічних досліджень, у ході яких оцінювали його вплив на функцію та електричну стабільність ішемізованого й декомпенсованого міокарда. Узагальнений аналіз результатів таких досліджень демонструє здатність цього засобу обмежувати зону некрозу при інфаркті міокарда, його антиішемічний та антиаритмічний ефекти, спроможність підвищувати систолічну функцію ЛШ у пацієнтів із СН [60].
За даними метааналізу, що охопив 32 клінічні дослідження екзогенного фосфокреатину в пацієнтів із ішемічною хворобою серця, ХСН та при кардіохірургічних втручаннях (загалом 3629 хворих), його застосування було асоційоване зі зниженням загальної смертності на 29%, а в рандомізованих дослідженнях за участю пацієнтів із ХСН – на 60% [66].
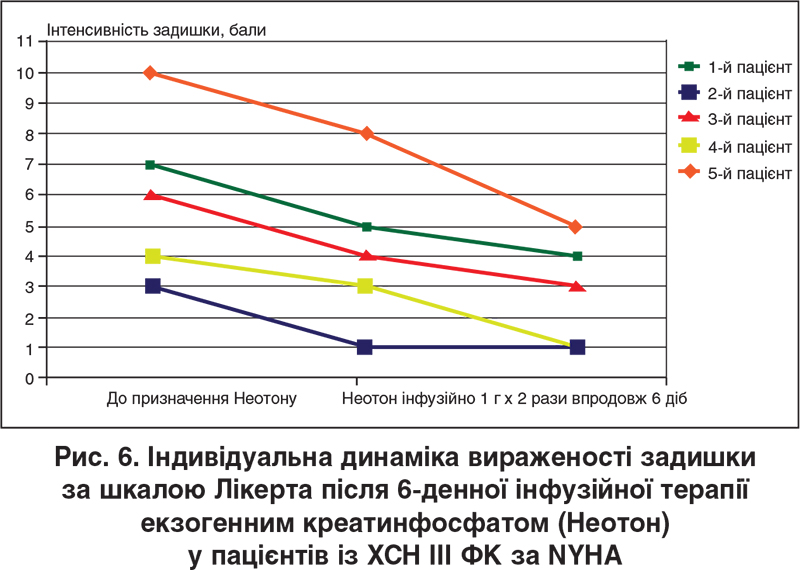
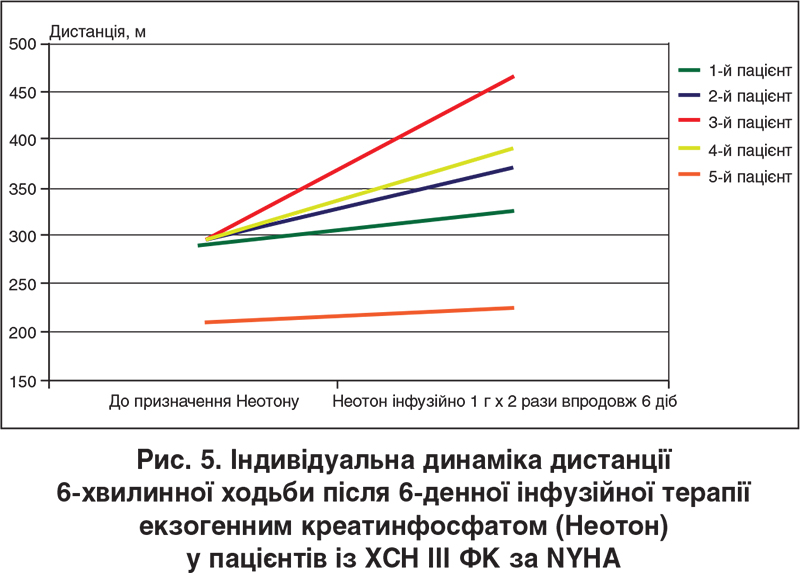
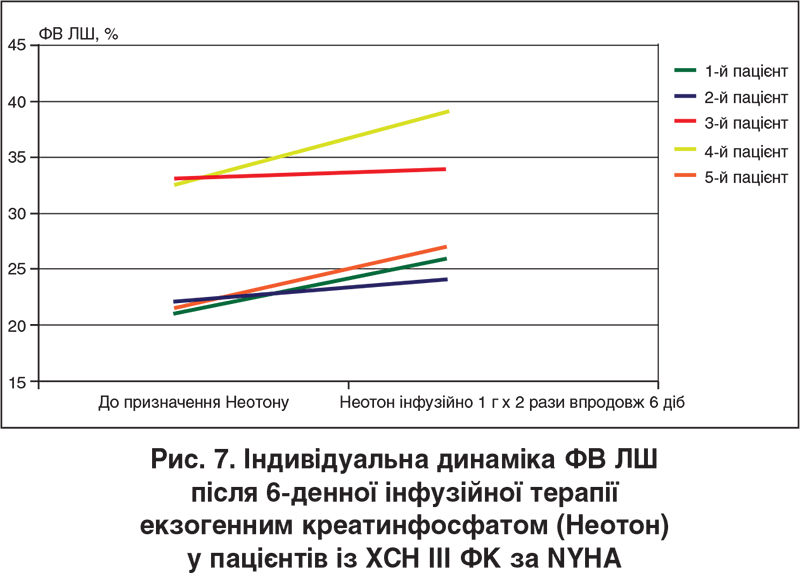
Наші клінічні спостереження, котрі є частиною започаткованого пілотного дослідження екзогенного фосфокреатину (Неотон) при ХСН зі зниженою ФВ ЛШ, продемонстрували, що внутрішньовенне інфузійне введення цього засобу в дозі 1000 мг кожні 12 год упродовж 6 діб пов’язане зі збільшенням дистанції ходьби за даними 6-хвилинного тесту та зменшенням вираженості задишки за шкалою Лікерта в усіх обстежених пацієнтів (рис. 5, 6), збільшенням витривалості м’язів-розгиначів нижніх кінцівок за даними відповідного стандартизованого тесту, а також зростанням ФВ ЛШ (у чотирьох випадках із п’яти), що показано на рисунку 7 (ехокардіографічні дані отримані разом із професором О.Г. Несукай та Й.Й. Гірешем). Зазначені попередні дані вбачаються обнадійливими з точки зору перспектив подальшого покращення функціональних можливостей пацієнтів із систолічною ХСН на тлі отримання раніше підібраної стандартної фармакотерапії (інгібітор АПФ / бета-блокатор / антагоністи мінералокортикоїдних рецепторів / діуретик).
Отже, науковий пошук щодо з’ясування механізмів прогресування периферичної міопатії при ХСН та розроблення нових засобів терапевтичного впливу на цей синдром наразі залишаються надзвичайно актуальними, відтак існує потреба в плануванні проведення нових досліджень у цьому напрямі.
Література
1. Kemppainen J., Fujimoto T., Kalliokoski K.K., et al. Myocardial and skeletal muscle glucose uptake during exercise in humans // J. Physiol. – 2002. – Vol. 542. – P. 403-412.
2. Schiaffino S., Reggiani C. Molecular diversity of myofibrillar proteins: gene reculation and functional significance // Physiol. Rev. – 1996. – Vol. 76. – P. 371-423.
3. Sullivan M.J., Green H.J., Coob F.R. Skeletal muscle biochemistry and histology in ambylatory patients with long-term heart failure // Circulation. –1990. – Vol. 81. – P. 518-527.
4. Lindsay D., Lovegrove C., Dunn M., et al. Histological abnormalities of diaphragmatic muscle may contribute to dyspnoea in heart failure // Circulation. – 1992. – Vol. 86. – 515A.
5. Le Jemtel T.H., Mancini D.M. Alterations in diaphragmatic and skeletal muscle in heart failure // In.: Heart Failure. – Ed.: Mann D.L. Second edition. – 2011. – P. 300-311.
6. Conzalez-Alonco J., Colbert J.A. Reductions in systemic and skeletal muscle blood flow and oxygen delivery limit maximal aerobic capacity in humans // Circulation. – 2003. – Vol. 107. – P. 824-830.
7. Clark A.L., Volterrani M., Swan J.W., et al. Leg blood flow, metabolism and exercise capacity in chronic heart failure // Int. J. Cardiol. – 1996. – Vol. 55 (2). – P. 127-135.
8. Cohen-Solal A., Logeart D., Guiti C., et al. Cardiac and peripheral responses to exercise in patients with chronic heart failure // European Heart J. –1999. – Vol. 20. – P. 931-945.
9. Katz S.D., Biasucci L., Sabba C., et al. Impaired endothelium-mediated vasodilation in the vasculareture of patients with congestive heart failure // J. Am. Coll. Cardiol. – 1992. – Vol. 19. – P. 918-925.
10. Le Jemtel T.H., Mascin C.S., Lucido D., et al. Failure to augment maximal limb blood flow in response to one-leg exercise in patient with severe heart failure // Circulation. – 1986. – Vol. 74. – P. 245-251.
11. Scheuermann-Freestone M., Madsen P.L., Manners D., et al. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes // Circulation. – 2003. – Vol. 107. – P. 3040-3046.
12. Ільницька М.Р. Інсулінорезистентність при хронічній серцевій недостатності (огляд літератури та власні дані) // Серцева недостатність. –2015. – № 3. – С. 6-10.
13. Chati Z., Zannad F., Jeandel C., et al. Physical deconditioning may be a mechanism for the skeletal muscle energy phosphate metabolism abnormalities in chronic heart failure // Am. Heart J. – 1996. – Vol. 131. – P. 560-566.
14. Mancini D.M., Coyle E., Coggan A., et al. Contribution of intrinsic skeletal muscle changes to 31P-NMR skeletal muscle metabolic abnormalities in patients with chronic heart failure // Circulation. – 1989. – Vol. 80. – P. 1338-1346.
15. Schaufelberger M., Andersson G., Eriksson B.O., et al. Skeletal muscle changes in patients with chronic heart failure before and after treatment with enalapril // European Heart J. – 1996. – Vol. 17. – P. 1678-1685.
16. Mettauer B., Zoll J., Sanchez H., еt al. Oxidative capacity of skeletal mescle in heart failure patients versus sedentary or active control subjects // J. Am. Coll. Cardiol. – 2001. – Vol. 38. – P. 947-954.
17. Drexler H., Riede U., Munzel T., et al. Alternations of skeletal muscle in chronic heart failure // Circulation. – 1992. – Vol. 85. – P. 1751-1759.
18. Sullivan M.J., Green H.J., Coob F.R. Skeletal muscle biochemistry in ambulatory patients with long-term heart failure // Circulation. – 1990. – Vol. 81. – P. 518-527.
19. Schaufelberger M., Eriksson B.O., Grimby G., et al. Skeletal muscle fiber composition and capillarization in patients with chronic heart failure: relation to exercise capacity and central hemodynamics // J. Card. Fail. – 1995. – Vol. 1. – P. 267-272.
20. Lunde P.K., Dahlstedt A.J., Bruton J.D., et al. Contraсtion and intracellular Ca2+ handing in isolated skeletal muscle of rats with congestive heart failure // Circ. Res. – 2001. – Vol. 88. – P. 1299-1305.
21. Бесага Є.М. Клінічні аспекти скелетної міопатії при хронічній серцевій недостатності // Укр. кардіол. журнал. – 2006. – № 4. – С. 58-62.
22. Opasich C., Ambrosino N., Felicetti G., еt al. Heart Failure-related myopathy Clinical and pathopysiological insights // Eur. Heart Failure. – 1999. – Vol. 20. – P. 1191-1200.
23. Бойцов С.А., Кириченко П.Ю., Пинегин А.Н. и соавт. Структурно-функциональное состояние поперечно-полосатой мускулатуры у больных с хронической сердечной недостаточностью различных функциональных классов // Сердечная недостаточность. – 2003. – Том 4, № 4. – С. 194-198.
24. Piepoli M., Clark A.L., Coats A.J.S. Muscle metaboreceptor in the hemodynamic, autonomic and ventilatory responses to exercise in men // Am. J. Physiol. – 1995. – Vol. 38 (4). – H1428-1436.
25. Ponikowski P.P., Chuna T.P., Francis D.P., et al. Muscle ergoreceptor overactivity reflects deterioration in clinical status and cardirespiratory reflex control in chronic heart failure // Circulation. – 2001. – Vol. 104. – P. 2324-30.
26. Oelberg D., Evans A.B., Hrovat M.I., et al. Skeletal muscle chemoreflex and pH in exercise ventilatory control // J. Appl. Physiol. – 1998. – Vol. 84. – P. 676-682.
27. Griendling K.K., Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling // Regul. Pept. – 2000. – Vol. 91. – P. 21-27.
28. Riede U.N., Forstermann U., Drexler H. Inducible nitric oxide synthase in skeletal muscle of patients with chronic heart failure // J. Am. Coll. Cardiol. – 1998. – Vol. 32 (4). – P. 964-969.
29. Hambrecht R., Adams V., Gielen S., et al. Exercise intolerance in patients with chronic heart failure and increased expression of inducible nitric oxide synthase in the skeletal muscle // J. Am. Coll. Cardiol. – 1999. – Vol. 33 (1). – P. 174-179.
30. Mann L.L. Activation of inflammatory mediators in heart failure // In.: Heart Failure. Ed.: D.L. Mann. Second edition. – 2011. – P. 163-184.
31. Caron M.A., Debigare R., Dekhuijzen P., et al. The respiratory muscles in chronic obstructive pulmonary disease: comparative assessment of the quadriceps and the diaphragm in patients with COPD // J. Appl. Physiol. – 2009. – Vol. 107. – P. 952-961.
32. Jankowska E.A., Bartosz B.B., Majda J., et al. Anabolic deficiency in men with chronic heart failure prevalence and detrimental impact on survival // Circulation. – 2006. – Vol. 114. – P. 1829-1837.
33. Fixler D., Atkins J., Mitchell J., et al. Blood flow to respiratory cardiac, and limb muscles in dogs during graded exercise // Am. J. Physiol. – 1976. – Vol. 231. – P. 1515-1519.
34. Lindsay D., Lovegrove C., Dunn M., et al. Histological abnormalities of diaphragmatic muscle may contribute to dyspnoea in heart failure // Circulation. – 1992. – Vol. 86. – P. 515A.
35. Tikunov B., Levine B., Mancini D.M. Chronic congestive heart failure elicits adaptations of endurance exercise in diaphragmatic muscle // Circulation. – 1997. – Vol. 95. – P. 910-916.
36. van Hees H., van der Heijden H., Hafmans T., et al. Impaired contractility and structural abnormalities in the diaphragm of congestive heart failure rats // Int. J. Cardiol. – 2008. – Vol. 128. – P. 326-335.
37. McParland C., Krishnan B., Wang Y., et al. Inspiratory muscle weakness and dyspnea in chronic heart failure // Am. Rev. Respir. Dis. – 1992. – Vol. 146. – P. 467-472.
38. Meyer F.J., Borst M.M., Zugck C., et al. Respiratory muscle dysfunction in congestive heart failure: clinical correlation and prognostic significance // Circulation. – 2001. – Vol. 103. – P. 2153-2158.
39. Mancini D.M., Henson D., LaManca J., et al. Respiratory muscle function and dyspnea in patients with heart failure // Circulation. – 1992. – Vol. 86 (Suppl. I). – I 515A.
40. Hulsmann M., Quittan M., Berger R., et al. Muscle strength as a predictor of long-term survival in severe congestive heart failure // Eur. J. Heart Failure. – 2004. – Vol. 6. – P. 101-107.
41. Besaga E. Knee flexors muscle strength is the predictor of long-term survival in heart failure // Eur. Heart J. – 2011. – Vol. 32 (Abstr. Suppl.): 611.
42. Vescovo G., Libera L.D., Serafini F., et al. Improved exercise tolerance after losartan and enalapril in heart failure: Correlation with changes in skeletal muscle myosin heavy chain composition // Circulation. – 1998. – Vol. 98. – P. 1742-1749.
43. Munzel T., Kurz S., Drexler H. Are alterations of skeletal muscle ultrastructure in patients with heart failure reversible under treatment with ACE inhibitors? // Herz. – 1993. – Vol. 18 (Suppl.). – P. 400-405.
44. Jondeau G., Katz S.D., Toussaint J.F., et al. Regional specificity of peak hyperemic response in patients with congestive heart failure: correlation with peak aerobic capacity // J. Am. Coll. Cardiol. – 1993. – Vol. 22. – P. 1399-1402.
45. McMurray J., Abdullah I., Dargie H.J., et al. Increased concentrations of tumor necrosis factor in «cachectic» patients with severe chronic heart failure // Br. Heart J. – 1994. – Vol. 15. – P. 1528-1532.
46. Ko D.T., Hebert P.R., Coffey C.S., et al. Adverse effects of β-blockers therapy for patients with heart failure // Arch. Intern. Med. – 2004. – Vol. 164 (13). – P. 1389-1394.
47. Амосова Е.Н., Руденко Ю.В., Андреев Е.В. и соавт. Преимущества контроля частоты сердечных сокращений с помощью комбинации ивабрадина с β-адреноблокатором по сравнению с полнодозовой терапией β-адреноблокатором во влиянии на толерантность к физической нагрузке, продольную систолическую и диастолическую функции миокарда и уровень NT-proBNP у больных ишемической болезнью сердца с умеренно сниженной фракцией выброса: результаты двухмесячного наблюдения // Серце і судини. – 2014. – № 1. – С. 19-26.
48. Christensen L.P., Zhang R., Zheng W., et al. Postmyocardial infarction remodeling and coronary reserve: effects of ivabradine and beta blockade therapy // Am. J. Physiol. Heart Circ. Physiol. – 2009. – Vol. 297. – P. H322-H330.
49. Yue T.L., Cheng H.Y., Lysko P.G., et al. Carvedilol, a new vasodilator and beta-adrenoreceptor antagonist, is antioxidant and free radical scavenger // J. Pharmacol. Exp. Ther. – 1992. – Vol. 263. – P. 92-98.
50. Вивчити клініко-патогенетичне значення та можливості корекції системних периферичних і метаболічних порушень при хронічній серцевій недостатності. Звіт про НДР (проміжний) / Кер. Л.Г. Воронков, відп. вик. Є.М. Бесага, Інститут кардіології ім. М.Д. Стражеска НАМН України. – К., 2004. – С. 43-51.
51. Anker S.D., Coats A.J., Rorcker E.B., et al. Does carvedilol prevent and reverse cardia cachexia in patients with severe heart failure? Results of the COPERNICUS study // Europ. Heart J. – 2002. – Vol. 4 (Suppl. Abstr.). – P. 394.
52. Воронков Л.Г., Богачева Н.В., Гавриленко Т.И., Корнилина Е.М. Клинико-фармакодинамические эффекты карведилола у больных с хронической сердечной недостаточностью // Матеріали об’єднаного пленуму правління Українських наукових товариств кардіологів, ревматологів та кардіохірургів з міжнародною участю «Серцева недостатність: сучасний стан проблеми». – К., 2002. – С. 40-41.
53. Radzewitz A., Miche E., Herrmann G., et al. Exercise and muscle strength training and their effect on quality of life in patients with chronic heart failure // Europ. J. Heart Failure. – 2002. – Vol. 4. – P. 627-634.
54. Erbs S., Hollriegel R., Linke A., еt al. Exercise training in patients with chronic heart failure (NYHA IIIb) promotes restoration of peripheral vasomotor function, induction of endogenous regeneration, and improvement of left ventricular function // Circ. Heart Failure. – 2010. – Vol. 3. – P. 486-494.
55. Taylor R.S., Sagar V.A., Davies E.J., et al. Exercise-based rehabilitation for heart failure // Cochrane Database Syst. Rev. – 2014. – Vol. 4. – CD003331.
56. Hambrecht R., Niebauer J., Fiehn E., еt al. Physical training in patients with stable chronic heart failure: effects on cardiorespiratory fitness and ultrastructural abnormalities of leg muscles // JACC. – 1995. – Vol. 25 (6). – P. 1239-1249.
57. Belardinelli R., Georgiou D., Scocco V., еt al. Low intensity exercise training in patients with chronic heart failure // J. Am. Coll. Cardiol. – 1995. – Vol. 26. – P. 975-982.
58. Belardinelli R., Scocco V., Purcaro A. Low intensity exercise training improves skeletal muscle oxidative capacity without changes in capillary growth in chronic heart failure // Circulation. – 1995. – Vol. 92. – P. 1-399.
59. Lenk K., Schur R., Linke A., et al. Impact of exercise training on myostatin expression in the myocardium and muscle in a chronic heart failure model // Europ. J. Heart Failure. – 2009. – Vol. 11. – P. 342-348.
60. Рудык Ю.С. Прямая коррекция энергетического статуса декомпенсированного миокарда у пациентов с тяжелой ХСН: в фокусе внимания – экзогенный фосфокреатин // Серцева недостатність. – 2016. – № 3. – С. 13-22.
61. Strumia E., Pelliccia F., D’Ambrosio G., еt al. Creatine phosphate: pharmacological and clinical perspectives // Adv. Ther. – 2012. – Vol. 29. – P. 99-123.
62. Weiss R.G., Gerstenblith G., Bottomley P.A. ATP flux through creatine kinase in the normal, stressed, and failing human heart // Proc. Natl. Sci. USA. – 2005. – Vol. 102. – P. 808-813.
63. Neubauer S., Horn M., Pabst T., еt al. Contributions of 31P-magnetic resonance spectroscopy to the understanding of dilated heart muscle disease // Europ. Heart J. – 1995. – Vol. 16. – P. 115-118.
64. Neubauer S., Horn M., Cramer M., et al. Myocardial phosphocreatine to ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy // Circulation. – 1997. – Vol. 96. – P. 2190-2196.
65. Adamopoulos S., Coats A.J., Brunnote F., еt al. Physical training improves skeletal muscle metabolism in patients with chronic heart failure // J. Am. Coll. Cardiol. – 1993. – Vol. 21. – P. 1101-1106.
66. Landoni G., Zangrillo A., Lomivorotov V. Cardiac protection with phosphocreatine: a meta-analysis // Interact. Cardio Vasc. Thorac. Surg. – 2016. – P. 1-10.
Журнал "СЕРЦЕВА НЕДОСТАТНІСТЬ та коморбідні стани" № 2, вересень, 2017 р.
