


31 жовтня, 2020
Мітохондріальні хвороби: генетична епідеміологія, діагностика та лікування
 В основу статті лягли результати досліджень колективу науковців і лікарів Харківського міжобласного спеціалізованого медико-генетичного центру – центру рідкісних (орфанних) захворювань, кафедри медичної генетики, Українського інституту клінічної генетики Харківського національного медичного університету та світовий досвід, відображений у публікаціях 2000-2020 рр.
В основу статті лягли результати досліджень колективу науковців і лікарів Харківського міжобласного спеціалізованого медико-генетичного центру – центру рідкісних (орфанних) захворювань, кафедри медичної генетики, Українського інституту клінічної генетики Харківського національного медичного університету та світовий досвід, відображений у публікаціях 2000-2020 рр.
Клінічна генетика на Слобожанщині заявила про себе появою медико-генетичного консультування сімей із гамартозами (факоматозами) на базі Харківської психіатричної клініки. Після 25 «років темряви» в українській клінічній генетиці у 1965 р. були досліджені характеристики спадкових порушень – муковісцидозу, адреногенітального синдрому. Так відроджувалася клінічна генетика у вигляді медико-генетичної консультації, яка сьогодні перетворилась у Харківський міжобласний спеціалізований медико-генетичний центр. Першому нашому пацієнтові сьогодні 50 років.
Сьогодення принесло людству безліч випробувань і безліч нових хвороб. Розлади аутистичного спектра (РАС) і рідкісні захворювання непомітно стали «тихою епідемією» сучасності. Потім «приєднався» синдром дефіциту уваги та гіперактивності (СДУГ). З’ясувалося, що 60% таких дітей мають ознаки мітохондріальних дисфункцій, а у 83% із них (за нашими даними) ці розлади асоційовані з прогресуючим перебігом цитомегаловірусної інфекції, здебільшого абдомінальної форми. Зараз уже відомо, що у 60% випадків СOVID-19 характеризується орально-фекальним типом зараження і перебігом із проявами мітохондріальної дисфункції. Абдомінальну вірусну хворобу (зумовлену цитомегаловірусом – ЦМВ, вірусами Епштейна – Барр – ВЕБ і простого герпесу) констатують у 86% дітей із РАС та СДУГ. А Peter J. McGuire, Eliza Gordon-Lipkin, Shannon Kruk, які вивчали роль мітохондріальної дисфункції та дисфункції мікробіоти в патогенезі COVID-19, в останніх роботах дійшли вкрай важливих для сучасної медицини висновків, що підтвердили отримані нами дані щодо ЦМВ і ВЕБ. Отже, мітохондрії є центром клітинного окисного гомеостазу; мітохондрії є основним джерелом активних форм кисню; позаклітинні мітохондрії містяться в крові, циркулюючих тромбоцитах і везикулах; патогенез COVID-19 посилюється гіперзапальним станом; запалення активує процеси, що призводять до окисного пошкодження мікробіоти та мітохондрій; пошкодження мітохондрій сприяє коагулопатії, фероптозу та мікробному дисбіозу; порушення функції мітохондрій крові та тромбоцитів може прискорити системну коагулопатію; орієнтація на дисфункцію мітохондрій може забезпечити корисні терапевтичні стратегії проти патогенезу COVID-19.
Інші медичні особливості часу характеризуються прогресуючим зростанням коморбідних форм захворювань, феноменом синтропії; поширенням стертих проявів рідкісних хвороб як наслідків взаємодії окремих генів і генетичних варіантів на тлі зміненого навколишнього середовища й епігенетичного статусу.
Безуспішне лікування традиційними препаратами психоневрологічних розладів у дітей зумовлює звертання сім’ї до клінічного генетика. Дедалі частіше саме РАС і СДУГ стають маркерами рідкісних хвороб, на тлі яких вони маніфестують, але здебільшого РАС і СДУГ мають мультифакторний характер. При направленні до генетичного центру зазначають такі діагнози, як затримка психомовленнєвого розвитку, порушення психомоторного розвитку, РАС і СДУГ. Діти переважно не мають стигм дисембріогенезу (за рідкісними випадками хромосомних захворювань і некласифікованих вад розвитку після екстракорпорального запліднення). Водночас при огляді відмічаються ознаки сполучнотканинної дисплазії, мітохондріальної (вторинної) дисфункції із м’язовою гіпотонією (плоско-вальгусна девіація, рідше – плоско-варусна). Основними скаргами є порушення психічного статусу із девіантною поведінкою, абдомінальними, імунними, психомоторними розладами.
При проведенні посиндромного аналізу все рідше знаходять конкретні синдроми, відомі в генетиці, здебільшого спостерігають індивідуальні комплекси широкого спектра ознак, зумовлені багатьма генами та генними поліморфізмами, спровоковані до маніфестації численними зовнішніми впливами (тригерами). Фенотип цих хворих ніби зітканий із багатьох перлин генних варіантів. Тому для клінічного генетика кожен хворий – це порушена система, а не окремі органи.
У зв’язку з цим роль клінічного генетика невпинно зростає як кермача команди 1+5: пацієнт + лікар-генетик + лікар-спеціаліст + лікар-біохімік + лікар-молекулярний генетик (біоінформатик) + фармакогенетик. Разом з тим зростає і його значення у розвитку сучасної медицини. Шляхи розвитку клінічної генетики в Україні пов’язані, безумовно, з її експансією у медицину. Це повинні зрозуміти всі – влада, суспільство, лікарі та пацієнти. Світ перебудовується на краще. Така перебудова еволюційна. Не випадково порушення енергетичного обміну «виросло» в мітохондріальну медицину.
У 1991 р. 8-й Міжнародний генетичний конгрес у Вашингтоні об’єднав 4 тисячі генетиків світу. Вперше проведено пленарне засідання, присвячене проблемі мітохондрій, уперше учасники конгресу почули доповіді про нову окрему гетерогенну групу хвороб зі значним клінічним поліморфізмом. Цю групу хвороб об’єднують єдині етіопатогенетичні фактори розвитку. Згодом було з’ясовано, що хвороби такого характеру частіше поширені як серед генетичних порушень із класичним типом успадкування, так і серед захворювань мультифакторного генезу. Ці захворювання характеризуються широким залученням клінічної патології, призводять до інвалідизації та вирізняються резистентністю до лікування. Така патологія включає велику кількість захворювань нервової системи, органа зору, м’язової та серцево-судинної систем, нирок, ендокринних органів. Ставало очевидно, що мітохондріальні захворювання можуть бути провідною причиною дегенеративних захворювань нервової системи.
Сьогодні на основі класичних і нових знань формується мітохондріальна медицина. Наш майже 30-річний досвід вивчення мітохондріальної патології дає підставу стверджувати, що мітохондріальна медицина зародилася ще в минулому столітті, а нині має наміри довести, що вона розвинулась на благо нашого життя та життя майбутніх поколінь.
Майже 90% всієї енергії, яка необхідна для клітин, тканин, органів та організму в цілому, забезпечується функціонуванням дихального ланцюга мітохондрій. Частина генів, причетних до мітохондріальної дисфункції (МТХД), міститься в ядрі клітини та успадковується однаковою мірою від обох батьків. Мітохондрії розміщені у цитоплазмі, мають власний спадковий апарат – мітохондріальну ДНК (мтДНК).
Уже відомі сотні мутацій і перебудов мітохондріального геному, асоційовані із широким спектром захворювань людини, що варіюють від летальних захворювань до нейродегенеративних порушень у різному віці. Незважаючи на велику кількість наукових досліджень, відсутнє розуміння мітохондріальної патології в цілому, яке б давало можливість своєчасно її діагностувати. Ці факти пов’язані з особливістю мітохондріального геному: йому притаманна більша швидкість мутагенезу порівняно із ядерним геномом. Так, точкові мутації в генах мтДНК виникають у 6-17 разів швидше, ніж в аналогічних генах нуклеарної ДНК.
Така особливість мтДНК разом із високою залежністю всіх органів і тканин від процесів синтезу молекули АТФ призвела до значного поширення мітохондріальних захворювань і зумовила роль генетичних дефектів мтДНК у патофізіологічних механізмах успадкованих нейродегенеративних захворювань, як і в процесах старіння.
Широкий спектр відкритих мітохондріальних мутацій, обмеженість популяційного складу сімей, у яких виявлені ці мутації, населенням Північної Америки, Західної Європи, України, Естонії, Росії, недостатня вивченість патогенного значення більшості мутацій, безумовно, затримують процес досконалого розуміння глибинної ролі мітохондріального геному у всіх проявах людського життя. Слід відмітити значний прогрес знань за останні 10-15 років, але вони ще не стали здобутком для лікарів у повсякденній праці. 2020 р. приніс нові дані: мітохондріальна медицина вже вийшла із неонатального періоду свого розвитку та охопила світ. Багато спеціалістів довели цей факт на віртуальному конгресі із мітохондріальної медицини EAPS 2020 (16-19 жовтня).
Слід відмітити, що вже на початку століття в різних науковців світу з’явилось розуміння важливості проведення детального скринінгу патогенних мтДНК мутацій у популяціях для визначення їх специфічності клінічному фенотипу, а також з метою розширення уявлення про вплив генетичного оточення мтДНК на експресію мітохондріальних захворювань. Ці дослідження здійснені відповідно до європейського проєкту INTAS за ініціативи професора Ріхарда Вільмса, який запросив нас до співпраці у молекулярній лабораторії Пенсильванського університету (професори Т. Шурр та С. Жаданов). Дані, отримані на першому етапі дослідження, дозволили нам вивчити генетичну епідеміологію МТХД в Україні (В. Гусар), запровадити діагностичний алгоритм для медико-генетичного консультування і розробити ефективні підходи до лікування та реабілітації пацієнтів із мітохондріальною патологією (О. Гречаніна). Визначена частка мітохондріальних захворювань у структурі метаболічних синдромів у населення України. Встановлено спектр мітохондріальних мутацій, які призводять до розвитку цієї патології в популяції (Д. Школьнікова). Впроваджено діагностичні алгоритми для проведення селективного генетичного скринінгу мітохондріальних захворювань. Результати такого скринінгу використані для прогнозування та профілактики, лікування і реабілітації пацієнтів із мітохондріальними захворюваннями. За цю серію робіт Президент України Леонід Кучма вручив колективу центру та кафедри 3 Державні премії України в галузі науки і техніки.
 Вручення Державної премії України в галузі науки і техніки (зліва направо) В. Гусар, Ю. Гречаніній і О. Гречаніній
Вручення Державної премії України в галузі науки і техніки (зліва направо) В. Гусар, Ю. Гречаніній і О. Гречаніній
Настав час, коли увага до МТХД стала вкрай необхідною, як і розуміння структури та функції «атомної електростанції клітини» – мітохондрій. Мітохондрії – це «енергетична станція» організму, яка виробляє велику кількість енергії у вигляді АТФ, зберігає кальцій для передачі сигналів клітинам, генерує тепло, опосередковує ріст і загибель клітин. Мітохондрії мають округлу чи овальну форму та розмір від 0,5 до 10 мкм. Кількість мітохондрій у клітині варіює. У людини еритроцити не містять мітохондрій, тоді як у клітинах печінки та м’язових клітинах їх сотні чи навіть тисячі. мтДНК сприйнятлива до мутацій, тому що не має надійного механізму репарації ДНК, притаманного ядерній ДНК. Відомо 229 генних дефектів мітохондріального енергетичного метаболізму, із них 192 ядерних, 37 мітохондріальних.
Розділяють два поняття – «мітохондріальна дисфункція» і «мітохондріальні захворювання». Мітохондріальна дисфункція – типовий патологічний процес, для якого не існує нозологічної й етіологічної специфічності. Слід вважати мітохондріальну дисфункцію новим патобіохімічним механізмом нейродегенеративних розладів широкого спектра (В.С. Сухоруков, 2007; І.Ф. Беленичев, В.І. Чернія, 2010).
Мітохондріальні хвороби – гетерогенна група захворювань, зумовлених генетичними, структурними, біохімічними дефектами мітохондрій і порушенням тканинного дихання.
За умови кодування білків мітохондріальним геномом мітохондріальні порушення успадковуються по материнській лінії, водночас зустрічається також і спорадичне, і соматичне успадкування. Відомо, що >98% білків, які залучені до мітохондріальних порушень, кодуються ядерним геномом. При мітохондріальних порушеннях, викликаних нуклеарним геномом, спостерігаються усі види успадкування – аутосомно-рецесивне, аутосомно-домінантне, Х-зчеплене, спорадичне, соматичне.
Мітохондріальні порушення ДНК мають свої особливості: не тільки гетероплазмію (кількість мутацій може бути різною), а й різний поріг клінічних ознак для різних органів, мультифакторні ураження (модифікація на ядерному фоні). Визначено особливості нуклеарних мутацій у ДНК, які стосуються мітохондрій: наявність ізоформи, тканиноспецифічних поєднань; Х-інактивація; мутації, які уражають мітохондріальну реплікацію ДНК.
В останні роки встановлено, що мітохондріальні мутації залежать від метаболітів, кофакторів, а раціон харчування відіграє значну роль у їх виникненні. Поширення ознак уражень енергетичного обміну у пацієнтів із різними формами спадкових і неспадкових захворювань показало необхідність розмежування клінічних понять – мітохондріальне захворювання та мітохондріальна дисфункція.
МТХД у сучасному розумінні – неспецифічний патологічний процес, що може виникнути при різних видах захворювань, до яких призводять різні патогенетичні фактори, і цей процес стає невід’ємною частиною патології. Оскільки від узгодженої роботи мітохондріальних ферментів залежить все енергопостачання клітини, будь-який дефект однієї з ланок цього ланцюга буде призводити до енергетичної недостатності (недостатності продукції АТФ у циклах клітинного дихання). Чим більше залежні тканинні елементи органів від аеробного дихання, тим вища його уразливість. Головні біохімічні процеси, розлади яких можуть призводити до розвитку клінічної картини мітохондріальних порушень, відбуваються в циклі трикарбонових кислот (цикл Кребса), карнітиновому циклі, при окисненні жирних кислот, транспорті електронів у дихальному ланцюгу й окисному фосфорилюванні. Нині спектр мітохондріальних порушень, пов’язаних із різними патологічними станами, розширюється – доведений внесок окиснення жирних кислот у розвиток гіпоглікемії у дітей, у синдром раптової смерті дитини.
Первинні МТХД мають свій особливий тип успадкування – материнський (цитоплазматичний, неменделівський). Первинні МТХД – це порушення ферментів або ферментних комплексів, безпосередньо залучених у вироблення хімічної енергії за допомогою окисного фосфорилювання: піруват-дегідрогеназний комплекс, дихальний ланцюг і АТФ-синтаза (R.J. Rodenburg, 2011; A. Garcia-Cazorla et al., 2006).
З точки зору клінічних ознак, патофізіології та генетики між окремими порушеннями існує значне перекривання: так, наприклад, у біосинтезі деяких білків беруть участь різні ферментні комплекси, а накопичені метаболіти можуть інгібувати інші ферменти.
МТХД класифікуються за типом мутацій. Чим більше накопичується мутацій у мтДНК, тим тяжчий перебіг захворювання. З еволюційної точки зору мутації можуть бути патогенними, слабопатогенними та нейтральними (W.C. Copeland, 2008; M.A. Huynen, 2009). Патогенні мутації знижують адаптацію їх носіїв, а потім швидко втрачаються в геномі. Висока швидкість мутування мтДНК є матрицею для їх незалежного виникнення на гетерогенному генетичному фоні, тому що дефектні мітохондрії з хронічною інтоксикацією вільними радикалами кисню проліферують швидше за нормальні, компенсуючи недостатність енергії (N. Shinohara et al., 2012).
У 1992 р. Wallace запропонував класифікацію мітохондропатій залежно від характеру мутацій:
- місенс-мутації (нейроофтальмопатія Лебера, пігментний ретиніт);
- мутації у генах тРНК (синдром MERRF, синдром MELAS);
- делеції або дуплікації ділянок мтДНК (зовнішня офтальмопатія; синдром Кернса – Сейра; синдром Пірсона; асиметричний птоз; двобічний птоз, поєднаний з офтальмопарезом та слабкістю м’язів нижніх кінцівок; дилатаційна кардіоміопатія; NARP-синдром, проксимальна міопатія з фокальною деплецією мітохондрій);
- мутації, що знижують число копій мтДНК (летальна інфантильна дихальна недостатність; синдром молочнокислого ацидозу);
- мутації в ядерній ДНК (фумарова ацидемія; глутарова ацидемія; дефіцит ацил-СоА-дегідрогенази жирних кислот із дуже довгим вуглецевим ланцюгом; дефіцит 3-гідроксіацил-СоА-дегідрогенази жирних кислот із довгим вуглецевим ланцюгом; дефіцит ацил-СоА-дегідрогенази жирних кислот із середнім вуглецевим ланцюгом; дефіцит ацил-СоА-дегідрогенази жирних кислот із коротким вуглецевим ланцюгом; підгостра некротизуюча енцефаломієлопатія Лея; прогресуюча склерозуюча поліодистрофія Альперса; трихополідистрофія Менкеса).
Існує і спрощена класифікація мітохондропатій: змістові заміни в структурних генах; мутації в генах рибосомних і транспортних РНК; структурні перебудови великих сегментів мтДНК, ОХРНОS. Остання класифікація достатньо широко використовується, і ми вважаємо її найбільш лаконічною та змістовною.
Порушення функції комплексу дихального ланцюга об’єднує велику кількість нозологічних форм. Функція комплексу І полягає у транспорті електронів від NADH до CoQ; CoQ дифундує до комплексу ІІІ. Кофактором у цьому ланцюзі є флавін (M. Nautiyal et al., 2010). Ферментом комплексу І є NADH-CoQ редуктаза. Цей комплекс найбільший у респіраторному ланцюзі: він складається із 46 протеїнів і 7 мітохондріальних білків. Порушення комплексу І призводить до енцефаломіопатії, кардіоміопатії, синдромів Лея, Барта та ін.
Мутації мтДНК у комплексі І зустрічаються при синдромах MELAS (tRNALeu), міопатії (tRNALeu), Лебера (LHON) (tRNALys), MERF (tRNALys), Leigh (ND4), діабеті та синдромі Фанконі (множинні делеції), атаксії, гіпогонадизмі (множинні делеції). Порушення комплексу І можуть бути викликані також мутацією ядерної ДНК. У такому випадку захворювання відрізняється чітким аутосомно-рецесивним успадкуванням, клінічними проявами енцефалопатії, кардіоміопатії, макроцефалії та лейкодистрофії, наявністю багатьох порушень, викликаних змінами у комплексі І (И.В. Леонтьева, 2006; P.F. Chinnery, 2009; M.A. Johnson et al., 2000).
Комплекс ІІ представлений ферментами сукцинатдегідрогеназою – CoQ-оксиредуктазою, у ньому сукцинатоксидаза перетворюється у фумарат, здійснюється перенесення електронів до убіхінону (CoQ). Цей комплекс є єдиним, який кодується у ядрі. Його кофактором є флавін і залізо-сульфатний протеїн. З порушенням комплексу ІІ пов’язані синдроми Кернса – Сейра, Лея, лейкоенцефалопатія, феохромоцитома, парагангліома (P.F. Chinnery, 2009; M.A. Johnson et al., 2000; D.C. Wallace, 2005; C.R. Roe et al., 2009).
Ензимом комплексу ІІІ є цитохром-С-редуктаза, яка складається із 10 нуклеарних субодиниць та 1 мітохондріальної, має білок, що є компонентом суперкомплексу І, ІІІ та ІV, в ньому електрони переносяться від CoQ до цитохрому С. Мітохондріальні дефекти представлені випадками синдрому Лебера (LHON), кардіоміопатією (P. Benit, 2009).
Комплекс IV складається із 10 нуклеарних та 3 мітохондріальних субодиниць. Його активним ензимом є цитохром-С-оксидаза. Мітохондріальні мутації у цьому комплексі призводять до проявів синдрому Лебера, енцефалопатії, міоглобулінурії, порушень слуху, нуклеарні мутації – до синдрому Лея, лейкодистрофії та енцефалопатії (L.J. Jacobs et al., 2006).
Комплекс V (фермент – АТФ-5-синтаза) складається із 16 нуклеарних та 2 мітохондріальних субодиниць. Цей комплекс здійснює синтез АТФ, його дефіцит призводить до неонатальної гіпотонії, кардіоміопатії, лактатацидозу (R.K. Nakamoto, 2008; J. Smet et al., 2009).
Комбінація дефіциту комплексів І, ІІІ, ІV та V викликається мутаціями мтДНК. Всі ці комплекси, включаючи і комплекс ІІ, складають мітохондріальний протеїн. Делеції мтДНК пошкоджують всі комплекси.
Таким чином, завдяки численним дослідженням сформовані загальні діагностичні маркери мітохондріальних хвороб: ураження багатьох органів, високий рівень молочної кислоти, ураження м’язів, очей, ендокринної системи, порушення рухів, гастроінтестинальні ураження (MNGIE; S. DiMauro, 2006).
Результати досліджень останніх років свідчать про різноманітність фенотипу однієї і тієї ж мутації. Так, мутація А3243G може бути асоційована з класичним варіантом MELAS (Н.Г. Даниленко, 2008; K.K. Abu-Amero et al., 2009) та з іншими формами МТХД.
У пацієнтів із мітохондріальними порушеннями як клінічні, так і клініко-біохімічні розлади є гетерогенними і часто неспецифічними. Симптоматика варіює залежно від віку дебюту (від народження до дорослого віку) і перебігу хвороби (швидкопрогресуюча, статична). У деяких пацієнтів буває уражена тільки одна тканина, тоді як інші мають мультисистемні порушення (D.C. Wallace, 2007; P.F. Chinnery, 2009).
У більшості пацієнтів м’язові та/або неврологічні порушення є основними симптомами, що виявляються в даний час. Деякі симптоми залежать від віку (затримка розвитку в неонатальному віці і непереносимість фізичних навантажень у дорослому віці), тоді як інші симптоми (гіпотонія, розумова відсталість) можуть виявлятися в будь-якому віці (A. Sanz, 2008).
Мітохондріальні хвороби можуть маніфестувати різними ознаками, в різному органі та в різному віці (P.F. Chinnery et al., 2000; P.F. Chinnery, S. DiMauro, 2006). У неонатальний період маніфестують синдром мітохондріальної деплеції (МDS), синдроми MELAS та NARP, хоча останні частіше зустрічаються у підлітків. Такі синдроми, як діабет із глухотою (MIDD), синдром Лебера, хронічна прогресуюча зовнішня офтальмоплегія (СPEO), мітохондріальна нейрогастроінтестинальна енцефаломіопатія (MNGIE), маніфестують у дорослих і мають прогредієнтний перебіг, який триває роками (P.F. Chinnery, 2009). Дані сучасної літератури і наш досвід свідчать про існування провокуючих факторів, загальних для всіх метаболічних хвороб, у тому числі і для мітохондріальних. До них належать інфекція, голодування, супер-стрес, травма, операція, переохолодження, прийом антибіотиків аміноглікозидного ряду, вальпроатів, антиретровірусних препаратів (A. Rezaie, 2007).
N. Blau (2010) систематизував різні ознаки мітохондропатій. Однак автор відзначив, що порушення у багатьох пацієнтів, особливо дітей, які задовольняють морфологічні, біохімічні та/або молекулярно-біологічні критерії, встановлені для мітохондріального порушення, не можна включити в жодну з вищезазначених категорій. Додаткове ускладнення полягає в тому, що у декількох пацієнтів відбувається поступова зміна клінічної картини з одного вираженого клінічного фенотипу на інший.
Найчастіші клінічні симптоми мітохондропатії (N. Blau, 2006): з боку центральної нервової системи (ЦНС) – напади судом, гіпотонія/гіпертонія, спастичність м’язів, транзиторний парапарез, летаргія/кома, затримка психомоторного розвитку/регресія, екстрапірамідні синдроми, атаксія (епізодична), порушення цілеспрямованих рухів, центральна гіповентиляція легенів, уповільнення/акселерація росту голови, сліпота, глухота (перцептивна); з боку скелетно-м’язової системи – непереносимість фізичного навантаження/легка стомлюваність, м’язова слабкість; з боку серця – кардіоміопатія (гіпертрофічна або дилатаційна), порушення провідності; з боку очей – птоз, обмежені рухи очей, страбізм, катаракта, пігментна дегенерація сітківки, атрофія зорового нерва; з боку печінки – печінкова недостатність; з боку нирок – дисфункція ниркових канальців; з боку ендокринної системи – цукровий і нецукровий діабет, затримка статевого дозрівання, гіпотиреоз, гіпопаратиреоз, екзокринна дисфункція підшлункової залози, дисфункція яєчників; з боку травного тракту – діарея (атрофія кишкових ворсинок), аномалія розвитку кишечнику; серед інших розладів – затримка розвитку, низькорослість, панцитопенія, анемія.
Таким чином, високий рівень клінічного поліморфізму та генетичної гетерогенності, різний вік маніфестації, здатність фенотипів мітохондріальних дисфункцій кардинально змінювати свій синдромальний характер ускладнюють діагностику МТХД, затримують початок лікування і реабілітації та негативно впливають на прогноз. Про це свідчать наші спостереження.
Під час проведення медико-генетичного консультування проводять аналіз родоводу. Його використовують для встановлення спадкового характеру ознаки, визначення типу успадкування (материнського, цитоплазматичного або ядерного, менделюючого). Оцінку родоводу здійснюють, щоб припустити орієнтовний процент гетероплазмії мтДНК на підставі вираженості клінічної симптоматики.
Діагностика мітохондріальних хвороб значно утруднена у зв’язку з поліорганністю ураження, великою кількістю неспецифічних ознак, що потребує виключення пошкоджень, викликаних неспадковими порушеннями.
Диференційна діагностика дає можливість за допомогою функціонально-діагностичних тестів звузити коло діагностичного пошуку мітохондріальних хвороб (С.И. Жаданов и др., 2006; С.Н. Иллариошкин, 2007; Н.Г. Даниленко, 2010). Більшість авторів вважає доцільним паралельно проводити дослідження показників метаболізму, насамперед лактату в плазмі крові та спинномозковій рідині (СМР). Визначення рівня лактату є першою ланкою в складній системі діагностики МТХД. Лактат накопичується у хворого унаслідок анаеробного розпаду глюкози.
Підвищений рівень лактату в СМР є важливим показником ураження ЦНС, проте він легко може викликатися стресом (страх венопункції), надмірними скороченнями м’язів (епілептичний стан), аноксією й іншими порушеннями. Достовірне значення дають лише результати, отримані на підставі двох або більше досліджень рівня лактату крові. У деяких пацієнтів з підтвердженим мітохондріальним дефектом не виявляють накопичення лактату в крові або сечі, проте в СМР цей показник часто підвищений. При деяких типах органічних ацидурій виділення лактату із сечею може бути підвищеним (Е.Д. Белоусова, 2008). Його рівень може підвищуватися при багатьох системних порушеннях під впливом накопичення токсинів. Тому гіпоксія, гіпотонія, кардіоміопатія, ниркова дисфункція, шок, сепсис супроводжуються підвищенням рівня лактату. Однак встановлено, що підвищення рівня лактату притаманне і деяким метаболічним захворюванням (органічні ацидурії, дефекти циклу Кребса, метаболізму пірувату, окиснення жирних кислот, метаболізму глікогену, дефекту біотинідази і тіаміну, порушенню комплексу окисного фосфорилювання в мітохондріях; C.M. Quinzii et al., 2008). Відомо, що у пацієнтів з ураженням ЦНС вміст лактату збільшується тільки у СМР (R.J. Rodenburg, 2011). Завдяки появі магнітно-резонансної спектроскопії (МРС) стало можливим визначення рівня лактату в мозку неінвазивним методом. Загалом встановлено, що підвищення рівня лактату є інформативним, але існує обмеження: при синдромах Кернса – Сейра, Лебера, MERRF його концентрація може бути незначно підвищена або нормальна (P.F. Chinnery et al., 2000). Підвищений рівень креатинкінази притаманний мітохондропатіям, дефектам окиснення жирних кислот і гліколізу.
Зміна рівня сечової кислоти має велике інформативне значення: підвищення свідчить про порушення обміну жирних кислот і мітохондріальну дисфункцію, а зниження – про порушення обміну пуринів або дефіцит кофактора молібдену. Зниження рівня міді притаманне хворобі Менкеса, церулоплазміну – хворобі Вільсона, Менкеса.
Гіпотиреоз та гіпопаратиреоз у поєднанні з іншими ознаками притаманні мітохондропатіям.
Особливе значення дослідники надають пошуку гострої метаболічної хвороби в неонатальний період у так званий безсимптомний інтервал, коли у малюків уже з другого дня життя розвивається м’язова гіпотонія, виникають проблеми з вигодовуванням, блювання, млявість, аномальне дихання, церебральні пароксизми. У таких випадках результати стандартних лабораторних досліджень найчастіше бувають нормальними або свідчать про інфікування (A. Jeyakumar et al., 2009).
Жодна з клінічних ознак не є специфічною або помітною. Підозра на те, що пацієнт має мітохондріальне порушення, виникає в тому випадку, коли доведена наявність щонайменше двох хронічних і нез’ясованих симптомів з вищезгаданого великого переліку, які переважно спостерігаються в двох не пов’язаних один з одним органах (Д.В. Влодавец и др., 2008).
Для діагностики мітохондріальних захворювань, на думку N. Blau (2010), необхідне проведення таких досліджень: визначення вмісту лактату і пірувату у крові, кетонових тіл у крові, амінокислот у крові та сечі, органічних кислот у сечі; комп’ютерна томографія (КТ) або магнітно-резонансна томографія (МРТ) чи МРС головного мозку. Слід підкреслити, що взяття зразків необхідно проводити у пацієнтів після прийому ними їжі (N. Blau, 2010; G.F. Hoffmann, 2011).
Деякі автори відзначають, що в одній або декількох біологічних рідинах більшості пацієнтів можна знайти накопичення метаболітів, асоційованих із мітохондріальним обміном (E. Holmes et al., 2006). Ми вважаємо, що в діагностиці мітохондропатій особливу увагу слід звернути на концентрацію лактату, бо після відновлення і трансамінування накопиченого пірувату утворюється надлишок лактату й аланіну.
Якщо на шляху окиснення пірувату зустрічається серйозна перешкода, і периферичні тканини не можуть адекватно видалити продукований лактат, то він накопичується в крові, сечі та/або СМР, залежно від ураженої тканини. Знижена активність дихального ланцюга проводитиме зміну рівноважного стану реакції лактатдегідрогенази в процесі перетворення пірувату в лактат. Тому у пацієнтів з дефектом дихального ланцюга має виявлятися підвищене відношення лактат/піруват у крові, тоді як недостатність піруватдегідрогенази повинна призводити до нормального відношення лактат/піруват. Проте цей механізм не у всіх випадках виявляється корисним для диференційної діагностики. Більше того, у деяких пацієнтів накопичення лактату в крові або сечі не відбувається.
Встановлено, що сучасний метод газової хроматографії дозволяє виявити відповідні метаболіти (A. Amirav et al., 2008). Корисним також може бути визначення концентрації й асоційоване з ним відношення кетонових тіл, ацетоацетату і 3-гідроксибутрату (E.Y. Plotnikov et al., 2009). У деяких пацієнтів з мітохондріальним порушенням спостерігаються кетоз і кетоацидурія. Іншим маркером мітохондріального дефекту може бути нефізіологічне підвищення рівня кетонових тіл після їди (В.В. Бережной и др., 2009; А.С Сенаторова, 2009). Підвищене відношення 3-гідроксибутират/ацетоацетат може свідчити про наявність дефекту в печінковій тканині дихального ланцюга. За світовими і нашими даними, діагностичну цінність має аналіз амінокислот у крові та сечі. При мітохондріальному порушені у багатьох пацієнтів відбувається підвищення рівня аланіну. На наявність дефекту дихального ланцюга може вказувати тяжка форма генералізованої амінацидурії, асоційованої з тубулопатією де Тоні – Дебре – Фанконі.
КТ і МРТ можуть надати важливу інформацію про локалізацію патологічних змін. Симетричні ураження в базальних ядрах і стовбурі головного мозку переконливо вказують на наявність синдрому Лея. Ми разом із З.З. Рожковою набули досвід виявлення підвищеного вмісту лактату у специфічних ділянках головного мозку за допомогою протонної МРС (J.A. da Rocha et al., 2008; R.P. Saneto, 2008).
У здорових людей швидкість кетогенезу, а отже – рівень ацетоацетату і 3-гідроксибутирату в крові після їди знижується, проте при мітохондріальних порушеннях він може підвищуватися (В.В. Бережной, 2009). Підвищений рівень аміаку в сироватці крові, підвищений рівень концентрації креатинкінази або білка в СМР не є показниками мітохондріального порушення. У разі виявлення порушення слід брати до уваги дефекти орнітинового циклу, цироз печінки, м’язову дистрофію або некроз головного мозку. Проте пацієнти із синдромами Кернса –Сейра і Лея часто мають підвищену концентрацію білка в СМР.
Своєчасна діагностика МТХД можлива на всіх етапах онтогенезу людини. Можливе і проведення пренатальної діагностики (Б.Ф. Ванюшин, 2006). Виконані нами попередні популяційні дослідження (О.Я. Гречаніна, Ю.Б. Гречаніна, В.А. Гусар, 2009) дозволили визначити мітохондріальні гаплотипи населення України для оцінки різноманітності, ступеня споріднення популяції і впливу генетичного фону на прояви мітохондріальних захворювань. Вперше охарактеризована структура мітохондріального генофонду населення України за даними поліморфізму нуклеотидних послідовностей, гіперваріабельний сегмент І (ГВС1) контрольного регіону і кодуючих регіонів мтДНК (рис. 1).
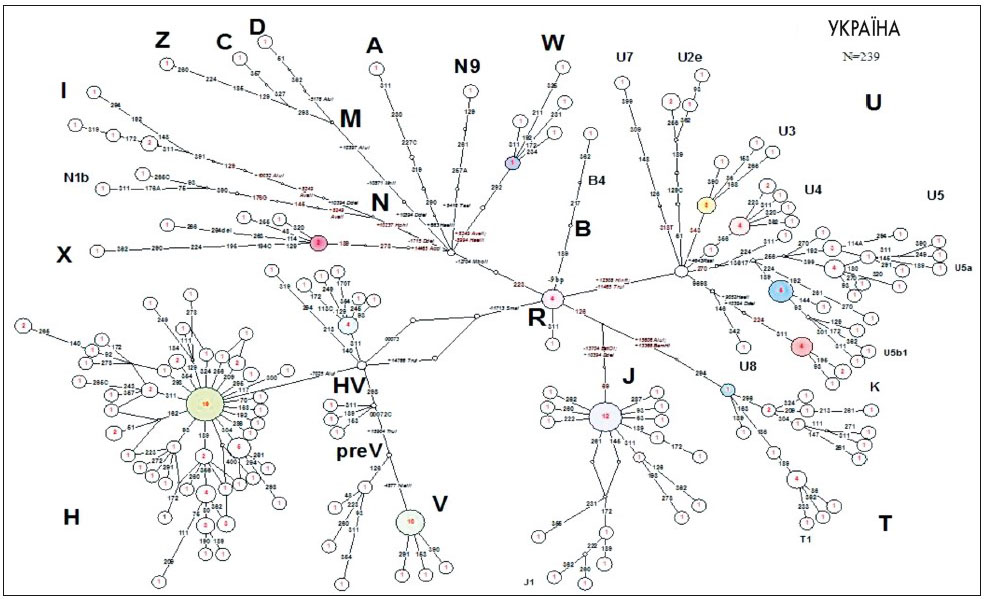
Рис. 1. Медіанна мережа, яка відображає філогенетичні відношення гаплотипів мтДНК у популяції українців
Ми оцінили також вплив популяційних поліморфізмів. У вибірці пацієнтів виявлено додаткові поліморфізми (генетичний фон) у гаплогрупах U5, T, X, які можуть відігравати важливу роль у проявах мітохондріальних захворювань (рис. 2).
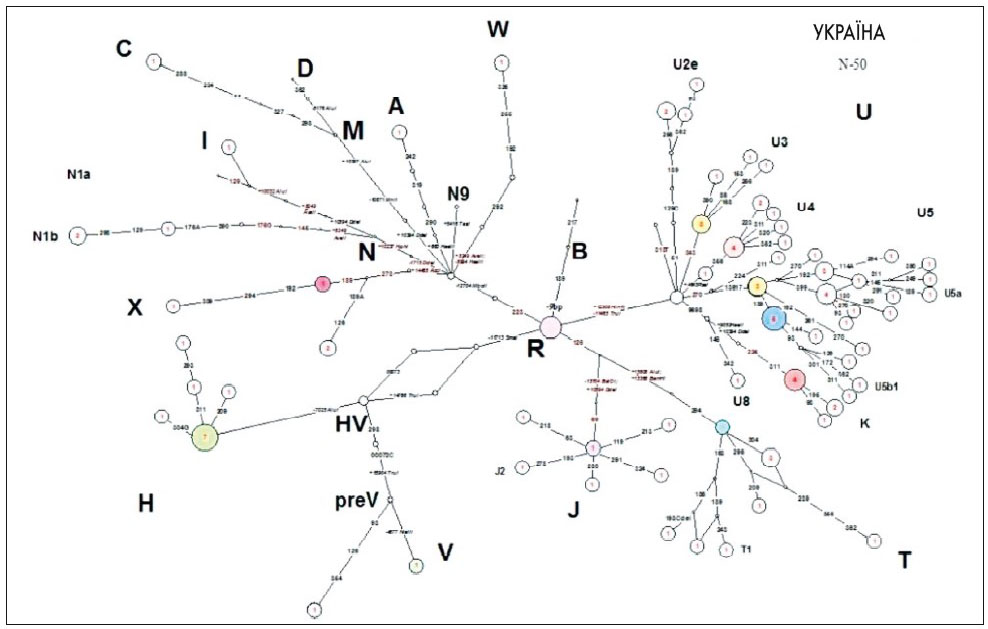 Рис. 2. Медіанна мережа, яка відображає філогенетичні відношення гаплотипів мтДНК у вибірці пацієнтів із клінічно встановленою мітохондріальною патологією
Рис. 2. Медіанна мережа, яка відображає філогенетичні відношення гаплотипів мтДНК у вибірці пацієнтів із клінічно встановленою мітохондріальною патологією
Отримані дані свідчать про генетичні особливості мтДНК у сучасній українській популяції, а також обґрунтовують необхідність їх обліку для ефективного прогнозування, діагностики та профілактики моногенної і мультифакторної патології, асоційованої з функціональною недостатністю мітохондрій.
Нам вдалося визначити вплив поліморфізмів мтДНК і поліморфних варіантів генів С677T MTHFR, А66G MTRR на клінічні прояви мітохондріальної дисфункції.
Концепція дослідження передбачала доведення запропонованого положення: вплив поліморфізмів мтДНК на вираженість МТХД відбувається внаслідок патологічного трансформування поліморфізмів мтДНК на тлі зміненого статусу метилювання як головного модифікатора геному та наявності тригера.
Метою роботи було розроблення нового напряму вивчення фундаментальних і прикладних проблем клінічного поліморфізму та генетичної гетерогенності мітохондріальної дисфункції, пов’язаної зі складною взаємодією популяційно-генетичних факторів, що здатні сформувати схильність до порушень енергетичного обміну на тлі зміненого епігенетичного статусу.
Були проведені молекулярно-генетичні дослідження; визначено частоту МТХД у структурі спадкових синдромів, вивчено клініко-генетичні особливості МТХД і патогенетичні механізми мітохондріальних енергетичних порушень; створено діагностичні алгоритми для проведення генетичного скринінгу МТХД у досліджуваній популяції (рис. 3).
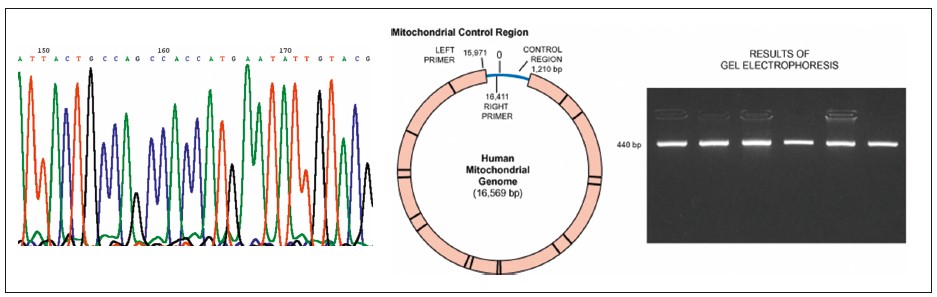
Рис. 3. Молекулярно-генетичні дослідження (фрагмент сіквенса ГВС1 та аналіз поліморфізму довжини рестрикційних фрагментів)
Вивчення поліморфізмів мтДНК дозволило встановити асоціації з патологією. Відмічене найчастіше включення в патологічний процес органів і систем при поліморфізмах тРНК-лізин: 8697G/A; 8860G; 8701G/А; 8856G/А; 8860А (CRS); 8251G/А; 8472С/Т; 8448Т/С; 8994G/А; 8337Т/С; 8794С/Т; 8584G/А; 8701А/G та при амінокислотній заміні тРНК-лізин (syn, thr/ala, pro/leu, met/val, met/thr, his/tyr, ala/thr), енцефалопатії асоційовані з тРНК-лізин і новими мутаціями (тРНК-лейцин; 3624А/G; 3594С/Т; 3705G/А; 3505А/G; 3552Т/А). Ураження м’язової, травної, серцево-судинної, ендокринної систем, органа зору частіше були асоційовані з поліморфізмами тРНК-лізин.
Доведена ймовірність залучення у патогенез МТХД не тільки специфічних для синдрому мутацій, а й генетичного оточення. При виявленій новій мутації фатального синдрому Лея 12706С гена ND5 у тканині мозку були присутні мутації F124L і Е145G ND5, які, ймовірно, стали причиною мутацій у зародкових клітинах матері та змінили, скоріш за все, функціонально важливі сайти, залучені в перенесення протонів, призвели до зміни протонного каналу і значно вплинули на фенотип синдрому Лея.
На підставі отриманих даних та з урахуванням клінічних критеріїв для діагностики мітохондріальних захворювань Niymegena (N.J. Wolf, J.A. Smeitinik, 2002) розроблено континуум клінічних ознак МТХД, генетичний навігатор (рис. 4), клінічний маршрут пацієнта з МТХД, алгоритм діагностики МТХД.
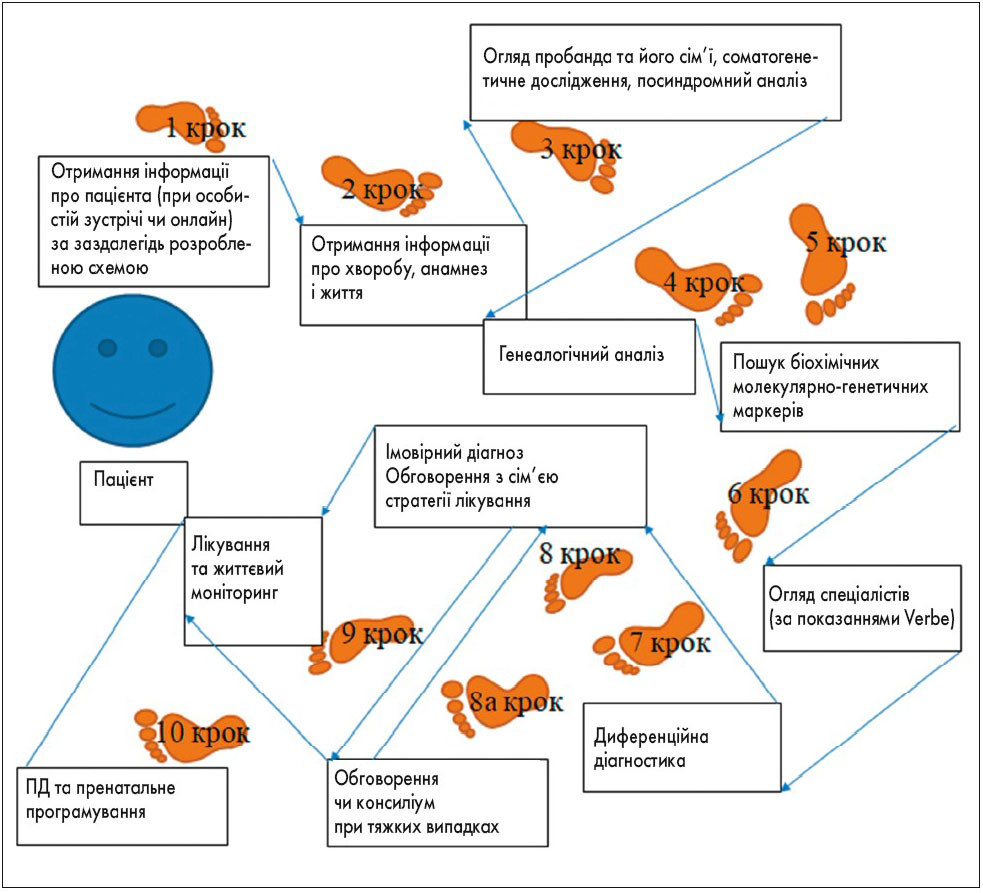 Рис. 4. Клінічний маршрут пацієнта з підозрою на МТХД – діагностична одіссея
Рис. 4. Клінічний маршрут пацієнта з підозрою на МТХД – діагностична одіссея
Всесвітнє товариство мітохондріальної медицини розробило мітохондріальні діагностичні критерії, які використовуються у процесі уточнювальної диференційної діагностики (N.J. Wolf, J.A. Smeitinik, 2002). Наш досвід свідчить про їх ефективність та інформативність, про доказовість результатів. Але у багатьох країнах ці критерії можуть бути технічно недоступними для використання у повному обсязі та дуже дорогими в плані досліджень для пацієнтів. Саме тому ми спростили для повсякденного використання алгоритм обстеження, виходячи з об’єктивних обставин. Але таке спрощення знижувало б доказовість висновків, і тому ми поділили цей процес на два рівні – базовий і додатковий із залученням різних лабораторій і відповідних закладів у світі.
Клінічні критерії
Нейром’язові прояви (максимум 2 бали):
а) прогресуюча зовнішня офтальмоплегія (2 бали);
б) птоз і гіпомімія (1 бал);
в) непереносимість фізичного навантаження, відмова від фізичних вправ (1 бал);
г) слабкість м’язів (1 бал);
д) рабдоміоліз (1 бал);
е) порушення на електроміограмі (1 бал).
ЦНС і залучення інших органів (максимум 2 бали):
а) ізольоване залучення ЦНС (1 бал);
б) залучення будь-яких інших ізольованих систем органів (1 бал);
в) залучення двох або більше систем органів (2 бали).
Метаболічні та візуальні дослідження (максимум 4 бали):
а) підвищений вміст лактату крові у 3 послідовних дослідженнях після їди (2 бали);
б) підвищений рівень лактату у СМР (2 бали);
в) підвищена концентрація аланіну крові (2 бали);
г) підвищений вміст аланіну у СМР (2 бали);
д) підвищений рівень трикарбоксильної кислоти у сечі (2 бали);
е) підвищення вмісту етилмалонової, 3-метилглутаконової чи дикарбоксильної кислоти (1 бал);
ж) виявлені на МРС порушення у м’язах зі зниженим відношенням фосфокреатину/Р (2 бали);
з) порушений сигнал Т2 у базальних ядрах при МРТ мозку (2 бали);
и) знижений обмін речовин у спокої чи дослідження фізичного навантаження (протокол велоергометрії; 2 бали).
Морфологія тканин (максимум 4 бали):
а) розірвані волокна при біопсії м’язів (2 бали якщо наявні, 4 бали якщо >2%);
б) дифузне зниження гістохімічної реакції цитохром-С-оксидази (4 бали);
в) виявлена гістохімічна позитивна реакція судин (сукцинатдегідрогеназа; 1 бал).
Підрахунок загальної кількості балів для оцінки клінічних критеріїв: точно – 8-12 балів; можливо – 5-7 балів; ймовірно – 2-4 бали; малоймовірно – 1 бал.
Біохімічні критерії
- Респірометричні порушення м’язів чи фібробластів (<5%). Оцінка комплексу V, з’єднання та виведення білка через мітохондріальну мембрану.
- Імунохімічне порушення OXPHOS чи імунофлуоресцентних показників біоптату скелетних м’язів (кількісний аналіз набору ферментів OXPHOS у тканинах біоптату). Порушення набору ферментів OXPHOS добре визначається за допомогою цього дослідження.
- Порушення (одиничне чи множинне) ферментів OXPHOS (вимірювання активності <5%). Дослідження має бути виконане на мітохондріях, виділених зі свіжої (не замороженої) тканини для мінімізації ризику артефактів, що викликані заморожуванням скелетних м’язів до виділення мітохондрій.
- Порушений кількісний вестерн-блотинг обраних одиниць OXPHOS з комплексу I-V (рівня <5%). Вестерн-блотинг може визначити порушення, не видимі у разі використання інших технологій.
- Порушений рівень CoQ10 (<50% контрольного значення). Оцінка первинних порушень синтезу коензиму Q10.
- Оцінка суперкомплексу: оптимальна функція OXPHOS потребує накопичення ферменту OXPHOS у суперкомплексах, що дозволяє ефективно формувати швидкий транспорт електронів. Суперкомплекси дають змогу ефективно створювати електрохімічний градієнт (протон) комплексами I, III, IV, який потім використовується комплексом V для синтезу АТФ. Утворення суперкомплексу порушується при різноманітних хворобах OXPHOS:
а) якщо порушене утворення суперкомплексу виявлене тільки при блакитному нативному OXPHOS імуноблотуванні – 0,5 бала, якщо в обох аналізах – 1 бал;
б) якщо порушене утворення монометричного ферментного комплексу наявне тільки у блакитному нативному OXPHOS імуноблотуванні – 0,5 бала, якщо в обох аналізах – 1 бал.
7. Порушена ферментна активність окисного фосфорилювання (кількісне визначення у гелі ферментної активності окисного фосфорилювання в інтактних ферментах. Це особливо важливо при визначенні активності АТФази комплексу V та асамблеї комплексу V.
Підрахунок загальної кількості балів для оцінки біохімічних критеріїв: малоймовірно – 0 балів; можливо >1; ймовірно =2; високоймовірно >2.
Генетичні критерії
- Деплеція мтДНК (число копій мтДНК <5%).
- Ідентифікація підтвердженої патогенної мтДНК чи нуклеарної мутації ДНК.
- Ідентифікація передбачуваної патогенної мтДНК чи мутації ядерної ДНК (нуклеарна). Мутація потребує додаткових даних, що підтверджують патологію.
- Мутація не виявлена.
Підрахунок балів для оцінки генетичних критеріїв: визначене захворювання – порушені 1 чи 2 критерії; можливе – порушений 3-й критерій; не визначене – наявність 4-го критерію.
Примітка. Критерій 4: невиявлення мутації не виключає мітохондріальної хвороби через велику кількість генів, асоційованих із мітохондріальною хворобою, і великою кількістю невиявлених генетичних асоціацій.
Нами розроблено й апробовано клінічний маршрут пацієнта з підозрою на мітохондріальну дисфункцію – діагностична одіссея (рис. 4). Наша одіссея відповідає діагностичному каскаду, запропонованому Johannes A. Mayr (рис. 5).
При пошуку ознак мітохондріальних порушень ми визначаємо ключові клінічні та біохімічні маркери МТХД: міопатичного синдрому (слабкість, гіпотонія, атрофія м’язів, зниження толерантності до фізичних навантажень); ураження центральної та периферичної нервової системи (респіраторний дистрес-синдром, порушення психомоторного розвитку, судоми, атаксія, пірамідні порушення, порушення зору – зовнішня офтальмоплегія, птоз тощо, полінейропатія). Не менш діагностично значущими були ознаки ураження печінки (прогресуюча гепатомегалія, фіброз, печінкова недостатність), серця (гіпертрофічна кардіоміопатія), нирок (тріада Фанконі – фосфатурія, глюкозурія, амінацидурія). Важливими ми вважаємо ендокринні порушення (затримка росту, гіпоглікемія), порушення слуху (нейросенсорна глухота), порушення зору (атрофія зорових нервів, пігментна дегенерація сітківки, катаракта), порушення шлунково-кишкового тракту (повторне блювання, діарея).
Метаболіти, досліджувані нами у пацієнтів з підозрою на МТХД, включали лактат, піруват, аланін, креатинін, гідроксибутират, ацетоацетат плазми/сироватки крові; лактат, аланін у СМР; метаболіти циклу Кребса, метилмалонову, 3-метилглутаконову й етилмалонову кислоти у сечі.
Додаткові дослідження включали ергометрію, спіроергометрію, МРС, навантажувальний тест глюкозою, аланіном.
Наші нижченаведені спостереження відображають хід уточнювальної діагностики та лікування.
Клінічний випадок 1
Пацієнт М-н. Синдром Лея.
Клінічні ознаки: неспокій дитини, розвиток із незначною затримкою. Збільшення симптоматики з 1 року 8 місяців після перенесеної інфекції, косоокість, плоско-вальгусна деформація стоп, невпевненість ходи, синдром рухових порушень, затримка мовленнєвого розвитку, спастичний трипарез, дизартрія, когнітивна недостатність, поліпшення на тлі енерготропної терапії.
На момент звернення дитині 3 роки: часті поперхування, порушення сну, немотивована посмішка, підвищене потовиділення.
У фенотипі: мармуровість шкірних покривів, поодинокі невуси, жорстке темне волосся, синофриз, довгі вії (національна особливість фенотипу), періорбітальні тіні, немотивована посмішка, косоокість, що розходиться, недостатньо розвинена підшкірна жирова клітковина, деформація грудної клітки.
Лабораторні показники: лактат ↑, співвідношення лактат/піруват ↑ – 22,6 (норма до 20), амінокислоти крові: ↑ триптофан, ↑ аргінін, ↑ лейцин, ↑ лізин, ↑ α-аміномасляна кислота, MTHFR 677CC (нормальна гомозигота), MTRR 66 AG (гетерозигота), MTR 2756 AA (нормальна гомозигота), В9 – норма, В12 – норма. Біохімічний аналіз крові: ↑ загальний холестерин. Мікроелементи: ↓ цинк, ↓ мідь. Вірусологічне обстеження: виявлено ВЕБ.
 Біопсія м’язів: мінімальні мітохондріальні аномалії (рис. 6). Спектроскопічний аналіз комплексів дихального ланцюга (2014 р.): мітохондріальна активність дихального ланцюга в нормі. Незначно знижений вміст лактатдегідрогенази.
Біопсія м’язів: мінімальні мітохондріальні аномалії (рис. 6). Спектроскопічний аналіз комплексів дихального ланцюга (2014 р.): мітохондріальна активність дихального ланцюга в нормі. Незначно знижений вміст лактатдегідрогенази.
Інструментальні дослідження: ультразвукове дослідження (УЗД) – реактивні зміни паренхіми печінки, помірна гепатомегалія; УЗД нирок – піелектазія зліва; електронейроміографія – ознаки грубого порушення проведення по кортико-спінальних шляхах. МРТ головного мозку (2014 р.): на рівні базальних ядер симетричні зони зміненого МР-сигналу (за рахунок геморагії або високого вмісту білка), вогнище високого сигналу та ділянки низького сигналу – ознаки ішемії. МР-картина структурних змін у проекції базальних ядер з обох боків і субепендимарно тілу лівого бічного шлуночка, імовірніше – за рахунок метаболічної (мітохондріальної?) енцефалопатії з ураженням підкіркової сірої речовини (MELAS?). Не можна виключити ЦМВ-інфекцію. Кіста прозорої перегородки. МРТ з негативною динамікою.
Синдром Лея зі спинномозковим ураженням.
Вивчення мітохондріальної ДНК: виявлена мутація 14487T>C гена MT-ND6, наявна в гетероплазмічному стані зі значенням 88%, що дає змогу прогнозувати протеїнові зміни pMt t63Val.
Секвенування за Сангером: виявлена патогенна мутація у мітохондріальному геномі (m.144887) у дитини та матері.
Клінічний випадок 2
Пацієнт М., 2006 р. н. Діагноз: MELAS-синдром (мутація А3243G). Порушення обміну сірковмісних амінокислот.
Клінічні ознаки: затримка розумового розвитку; напади тоніко-клонічних судом; емоційна лабільність; розгальмування; когнітивні порушення; не завжди виконує інструкції; періодичний біль у кінцівках; ходить навшпиньки. Хворий з народження. Народився в асфіксії в 35 тижнів – у матері прееклампсія. До 1 року тричі хворів на гострі респіраторні вірусні інфекції; анемія; гіпотрофія; тоніко-клонічні судоми; слабкість у ногах; часті падіння; пірамідна недостатність.
У 6,5 року: напад блювання; судоми; втрата свідомості; вторинна (токсична) кардіоміопатія; лямбліоз; ВЕБ; синдром мінімальної мозкової дисфункції; затримка психомоторного розвитку; епісиндром.
З 7 років: когнітивна епілептиформна дезінтеграція; психоорганічний синдром з інтелектуально-мнестичною недостатністю; порушення експресивного мовлення.
Молекулярне дослідження: полімеразна ланцюгова реакція, аналіз букального зішкрібу – виявлена мутація А3243П, що зумовлює розвиток синдрому MELAS. Кількість мутантних копій у гені лейцинової РНК складає понад 40%.
Лабораторні показники: ↑ лактат, ↑ гомоцистеїн, В9 – норма, В12 – норма, вірусологічне обстеження до капсидного білка та ядерного антигену ВЕБ – результат позитивний. Біохімічний аналіз крові: ↑ аспартатамінотрансфераза, ↑ сечовина, ↑ лактатдегідрогеназа, ↑ креатинкіназа. ГХ-МСС – виявлені зміни метаболітів: грибів, зокрема дріжджів, бактерій у травному тракті, кісткової тканини, нейротрансмітерів, зниження триптофану, кетоз, недостатність В6, В9, коензиму Q10.
Молекулярний аналіз: MTHFR 677CC (нормальна гомозигота), MTRR 66 AА (нормальна гомозигота), MTR 2756 AA (нормальна гомозигота); ВЕРХ амінокислот крові: ↓ метіонін, ↓ цистин, ↓ глутамін, ↓ цитрулін, ↓ треонін, ↓ гліцин, ↓ серин.
Інструментальні дослідження: електронейроміографія у 3 роки – змін не виявлено; МРТ головного мозку в 6,5 року – МР-ознак вогнищевого ураження паренхіми не виявлено; електроенцефалографія – пароксизмальна біоелектрична активність головного мозку; ехоенцефалоскопія – ознаки внутрішньочерепної гіпертензії; реоенцефалографія – порушений венозний відтік, асиметрія кровонаповнення; електрокардіографія – порушення процесів реполяризації міокарда у верхівково-бічній ділянці; функціональна кардіографія – зниження амплітуди I тону, фрагментований систолічний шум; УЗД щитоподібної залози – гіперплазія 1-2 ступеня; ехокардіографія – періодично мезосистолічні пролапси мітрального клапана 1 ступеня, фракція викиду – 64%, у лівому шлуночку – тонкі аберантні хорди. Рентгенографія попереково-крижового відділу хребта – перехідний попереково-крижовий хребець із явищами сакралізації, Spina bifida S1, S2. Нестабільність L2-L3-L4-L5.
Дитина перебуває на обліку в Харківському міжобласному спеціалізованому медико-генетичному центрі протягом 3 років, лікується відповідно до клінічного протоколу та додаткових призначень з урахуванням результатів досліджень із перманентним позитивним ефектом.
Лікування при МТХД. Основні вимоги: лікування має бути доказовим, раціональним, ефективним, безпечним, враховувати спектр застосовуваних препаратів та їх дії, а також біологічні властивості препаратів і їх компонентів, у тому числі взаємодії цих компонентів, без спрощення розуміння патофізіологічних процесів (табл.).
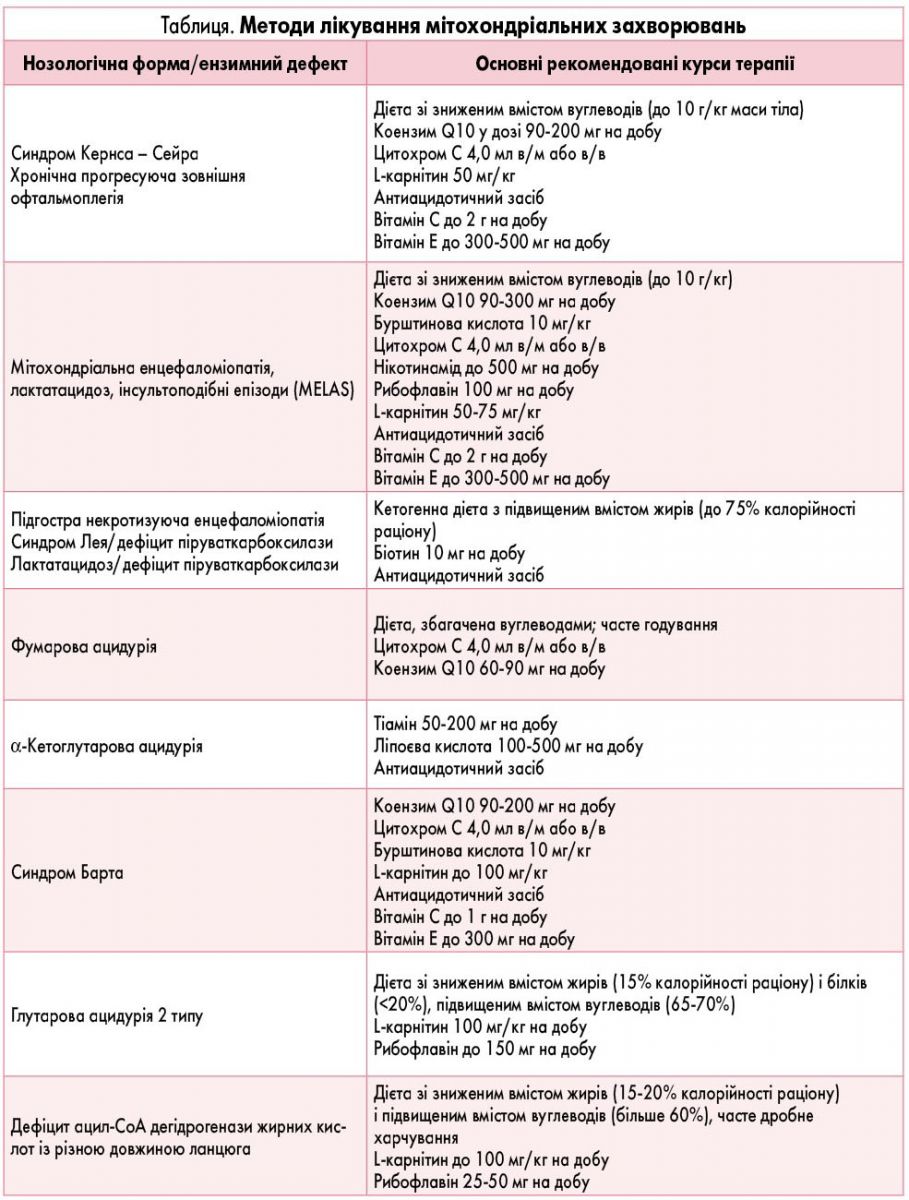
Сучасні підходи до ведення та лікування пацієнтів з мітохондріальними хворобами і мітохондріальною дисфункцією буде викладено в наступній публікації.
Вкрай необхідна термінова просвітницька робота серед населення і лікарів усіх спеціальностей із проблем мітохондріальної медицини, без якої жодні генетичні програми не дадуть очікуваного ефекту і не збільшать кількість хворих, які отримають адекватну медичну допомогу.
1 листопада у Харківському міжобласному спеціалізованому медико-генетичному центрі планується відкриття першої в Україні клініки мітохондріальної та епігенетичної медицини.
Тематичний номер «Педіатрія» №4 (55) 2020 р.
