


24 лютого, 2021
Хвороба Фабрі як специфічна причина інсульту в молодому віці
У кожного шостого пацієнта з інсультом він розвивається у віці від 18 до 50 років. При цьому в 15‑40% випадків причина ішемічного інсульту в молодих осіб залишається невстановленою. Хворобі Фабрі (ХФ) як одній із причин виникнення інсульту в молодому віці було присвячено вебінар, проведений 23 грудня 2020 року компанією ТОВ «Санофі-Авентіс Україна».
Фактори ризику інсульту в молодих осіб
 Завідувач кафедри нервових хвороб із курсом нейрохірургії Вінницького національного медичного університету імені М.І. Пирогова МОЗ України, д. мед. н., професор Сергій Петрович Московко виступив із доповіддю на тему «Фактори ризику інсульту в молодих осіб». Так, основними чинниками ризику виникнення інсульту в молодому віці є атеросклероз, кардіоемболізм, оклюзії судин, захворювання великих артерій, хвороба моямоя, дисекція інтракраніальних артерій, еклампсія; гематологічні захворювання, як-то ХФ, гомоцистинурія, синдром MELAS; мігрень, ВІЛ-інфекція, вагітність, приймання оральних контрацептивів тощо.
Завідувач кафедри нервових хвороб із курсом нейрохірургії Вінницького національного медичного університету імені М.І. Пирогова МОЗ України, д. мед. н., професор Сергій Петрович Московко виступив із доповіддю на тему «Фактори ризику інсульту в молодих осіб». Так, основними чинниками ризику виникнення інсульту в молодому віці є атеросклероз, кардіоемболізм, оклюзії судин, захворювання великих артерій, хвороба моямоя, дисекція інтракраніальних артерій, еклампсія; гематологічні захворювання, як-то ХФ, гомоцистинурія, синдром MELAS; мігрень, ВІЛ-інфекція, вагітність, приймання оральних контрацептивів тощо.
ХФ є Х-зчепленою рецесивною лізосомною хворобою накопичення, що виникає внаслідок дефіциту ферменту α-галактозидази та викликає ендотеліальну васкулопатію з подальшою церебральною ішемією. Науковці провели дослідження, в якому вивчали частоту нерозпізнаної ХФ у 721 пацієнта віком від 18 до 55 років, що переніс гострий інсульт. Було виключено осіб із факторами ризику інсульту (куріння, стенози сонних артерій, кардіоемболії, коагулопатії, ожиріння). В осіб з інсультом – 21 із 432 чоловіків та 7 із 289 жінок – було встановлено наявність біологічно значущої мутації в гені α-галактозидази. Таким чином, було продемонстровано високу частоту ХФ у когорті хворих після криптогенного інсульту, що відповідає приблизно 1,2% у молодих пацієнтів з інсультом (Rolfs et al., 2005).
Слід зазначити, що ХФ як специфічний чинник дуже важко діагностувати, насамперед тому, що це рідкісне генетичне захворювання. До того ж клінічна картина ХФ є поліморфною, а неспецифічні симптоми з боку деяких систем часто перекриваються із такими інших захворювань, як-то розсіяний склероз, гостра ревматична лихоманка тощо (Kubo, 2017).
Широкий клінічний поліморфізм, здатність ХФ маскуватися під інші поширеніші хвороби та низька проінформованість медичних працівників щодо спадкової патології часто є причиною того, що діагноз ХФ так і залишається невстановленим або відтермінованим. У середньому затримка зі встановленням діагнозу становить 8‑14 та 16‑19 років у чоловіків і жінок відповідно (через специфіку інактивації Х-хромосоми та мозаїцизм). Тому лікарям, зокрема сімейним, варто звертати увагу на родинний анамнез.
! За даними реєстру Фабрі, серед 2446 пацієнтів із ХФ інсульт трапився у 138 осіб, зокрема у 86 із 1243 чоловіків та 52 із 1203 жінок. Більшість інсультів були ішемічного типу (87%), середній вік першого епізоду в чоловіків та жінок становив 39,0 та 45,7 року відповідно (Sims et al., 2009).
Хвороба Фабрі: успадкування, типи та дебюти
 Яна Ігорівна Дороніна, лікарка-педіатриня центру орфанних захворювань Національної дитячої спеціалізованої лікарні «Охматдит» (м. Київ), розповіла про успадкування, типи та дебюти ХФ. Спікерка зауважила, що ХФ – одна з найчастіших форм спадкових ферментопатій, об’єднаних у загальну групу захворювань лізосомного накопичення, що має хронічний прогресувальний мультисистемний характер зі значним клінічним поліморфізмом та широким спектром мінливості симптомів. Одним із важливих симптомів, а інколи навіть єдиним, є цереброваскулярні прояви. Водночас, на відміну від інших спадкових хвороб, для цього захворювання доступні методи діагностики та ефективні схеми патогенетичного лікування, зокрема ферментозамісна терапія (ФЗТ), що понад 15 років представлена на фармринку України. Тому поінформованість клініцистів є запорукою повноцінного й успішного лікування та відповідної якості життя таких пацієнтів.
Яна Ігорівна Дороніна, лікарка-педіатриня центру орфанних захворювань Національної дитячої спеціалізованої лікарні «Охматдит» (м. Київ), розповіла про успадкування, типи та дебюти ХФ. Спікерка зауважила, що ХФ – одна з найчастіших форм спадкових ферментопатій, об’єднаних у загальну групу захворювань лізосомного накопичення, що має хронічний прогресувальний мультисистемний характер зі значним клінічним поліморфізмом та широким спектром мінливості симптомів. Одним із важливих симптомів, а інколи навіть єдиним, є цереброваскулярні прояви. Водночас, на відміну від інших спадкових хвороб, для цього захворювання доступні методи діагностики та ефективні схеми патогенетичного лікування, зокрема ферментозамісна терапія (ФЗТ), що понад 15 років представлена на фармринку України. Тому поінформованість клініцистів є запорукою повноцінного й успішного лікування та відповідної якості життя таких пацієнтів.
ХФ спричинена мутаціями в гені GLA, що призводять до повного чи часткового дефіциту лізосомної гідролази. Як наслідок, порушується катаболізм глікосфінголіпідів з їхнім відкладанням у клітинах різного типу – ендотелії, непосмугованій мускулатурі, фіброцитах, епітелії канальців нирок, нейронах, гангліях. Хвороба має Х-зчеплений рецесивний тип успадкування, тобто жінка із ХФ може передати її дітям незалежно від статі. Відповідно, хворий чоловік може передати захворювання лише донькам і ніколи – синам.
За даними Akhtar та Elliott (2018), залежно від фенотипових ознак, виділяють такі типи ХФ (співвідношення поширеності становить 1:7):
- класичний (тип 1) – характерні ранній початок (переважно у дитинстві), швидке прогресування, мультиорганність, ураження з типовими
- симптомами та ускладненнями;
- некласичний (тип 2, атиповий) – пізній початок (після 20 років), повільне прогресування, частіше моносистемний (умовно) варіант, як-от кардіальний, нирковий, інсультний тощо.
Жінки можуть мати тип 1 чи 2 та бути безсимптомними носіями. Тип 2 частіше маніфестує тяжчим кардіальним варіантом. Порівняно із чоловіками для жінок характерна більш пізня маніфестація (на 10‑15 років), можливі менш тяжка клінічна картина та повільніше прогресування; основна причина смерті – серцева недостатність (Lenders et al., 2016). Водночас ознаки захворювання дещо відрізняються залежно від статі. Зокрема, у чоловіків завжди наявні симптоми хвороби, тобто переважає тип 1. Тип 2 частіше маніфестує нирковим варіантом або ранніми інсультами. Клінічна картина більш виражена, а ознаки з’являються раніше, ніж у жінок. Основною причиною смерті є ниркова недостатність.
Ключовими ранніми клінічними симптомами у жінок та чоловіків є невропатичний біль (у 58,8 та 40,5% відповідно), гастроінтестинальні симптоми (у 23,2 та 11,4%), шкірні прояви (у 19,6 та 7,6%), а також кардіальні, ниркові та цереброваскулярні (Hopkin et al., 2008).
Першими та найчастіше інвалідизувальними проявами є неврологічні:
- акропарестезії;
- больовий криз (епізодичний або криз Фабрі);
- термолабільність і непереносимість спеки та/чи холоду;
- порушення потовиділення (гіпо- чи ангідроз);
- погіршення слуху (сенсонейронна втрата слуху).
Шкірні симптоми (у 66% чоловіків і 36% жінок) також є дебютними (1‑2-га декада життя) та проявляються у вигляді ангіокератом. Із часом вони поширюються тілом і можуть стати причиною кровотечі. Також важливими специфічними симптомами є офтальмологічні прояви (1‑2-га декада життя) – кератопатія (>70% випадків) – золотаво-коричнева або сіра опалесценція, катаракта й судинні зміни. Але найчастіше пацієнти дебютують шлунково-кишковими проявами (1-ша декада життя) у вигляді діареї та неспецифічних порушень (до 60% випадків), як-от нудота, блювання, закрепи, печія, здуття після їди, повільний приріст маси тіла.
Кардіальні симптоми можуть мати місце за класичного типу (2‑3-тя декада життя), а також бути єдиною ознакою атипового захворювання (4‑5-та декада) та проявлятися кардіоміопатією, клапанною недостатністю, субклінічними порушеннями: скороченням інтервалу PR, порушенням провідності, аритміями, розширенням QRS тощо. При цьому клінічними неспецифічними проявами є задишка, швидка стомлюваність, зменшення стійкості до навантаження, напади стенокардії. Також можливі такі відстрочені розлади, як фіброз серця та порушення серцевого ритму, кардіоміопатія з дилатацією лівого шлуночка (ЛШ).
Як за класичного типу, так і атипового перебігу можуть траплятися ниркові прояви, зокрема альбумінурія, сечовий синдром, а також відстрочені зміни (вторинний нефротичний синдром, інтерстиціальний нефрит).
ХФ належить до захворювань малих судин, що зумовлює високий ризик розвитку транзиторних ішемічних атак (ТІА) та ішемічних/геморагічних інсультів у молодому віці: частота інсультів становить 6,9% у чоловіків та 4,3% у жінок. У більшості хворих інсульт трапляється у віці 20‑50 років, зокрема в кожного 5-го з них – до 30 років. До того ж пацієнти, що не отримують патогенетичної терапії, схильні до рецидивів інсультних станів і ТІА (Sims et al., 2009). Варто зауважити, що у 50% випадків перенесений інсульт є дебютом ХФ, через що її слід підозрювати у всіх хворих із раннім розвитком інсульту навіть за відсутності очевидних причин та факторів ризику.
Таким чином, підозрювати ХФ слід при інсульті невідомої етіології в молодому віці (˂55 років), ангіокератомах, акропарестезіях, аритміях невідомої етіології, гіпертрофії ЛШ або гіпертрофічній кардіміопатії, гіпо- або ангідрозі, характерному «мутовчастому» помутнінні рогівки, нирковій недостатності невідомої етіології, а також у разі незрозумілої протеїнурії або мікроальбумінурії.
Також дуже важливим є сімейний скринінг у групах високого ризику. При цьому слід пам’ятати, що одна й та сама мутація у членів однієї родини може мати різні клінічні прояви.
Для наглядного прикладу нижче наведені клінічні випадки розвитку ХФ та аспекти ведення таких пацієнтів.
Клінічний випадок 1
Пацієнт Н., 14 років.
Скарги. Висипання на шкірі, біль у кінцівках, непереносимість спеки, зниження потовиділення, епізоди підвищення АТ та температури тіла, зниження соціальної активності та якості життя.
Анамнез. У 4 роки зниження потовиділення, непереносимість спеки, блідість та похолодіння нижніх кінцівок. У віці 10 років висипання на шкірі (червонуваті, з фіолетовим відтінком, не бліднуть під час натискання, 2‑3 мм у діаметрі), неприємні відчуття у дистальних відділах кінцівок з іррадіацією у проксимальні відділи, зі збільшенням інтенсивності при фізичних навантаженнях, кліматичних змінах, супутніх інфекціях із підйомом температури до субфебрильних значень та підвищенням артеріального тиску (АТ) до 140/90 мм рт. ст.
Електронейроміографія: ознаки помірно вираженої сенсорної поліневропатії нижніх кінцівок.
Анамнез життя: без особливостей. Генеалогічний аналіз: сестра (не хворіє на ХФ), мати померла від інфаркту у віці 60 років. Упродовж 10 років пацієнт перебував на обліку в педіатра, дерматолога, невролога, ендокринолога та мав робочий діагноз «синдром Рейно, поліневропатія нижніх кінцівок неясного ґенезу».
У 14 років у ЦОЗ НДСЛ «Охматдит» проведено специфічну діагностику ХФ: активність α-галактозидази в лейкоцитах крові – 7,8 мкм/л/год (референтні значення – 43‑67 мкм/л/год); молекулярно-генетичне обстеження: методом прямого автоматичного секвенування виявлено мутацію в гені GLA у гемізиготному стані, клас патогенності перший. ХФ підтверджено.
Обстеження. Загальний та біохімічний аналіз крові: без особливостей. Загальний аналіз сечі: без особливостей. Електрокардіографія: синусова дихальна аритмія. Ехокардіографія (ЕхоКГ): товщина задньої стінки ЛШ (ТЗСЛШ) – 12 мм, товщина міжшлуночкової перегородки в діастолу (ТМШП) – 12 мм. Ультразвукове дослідження органів черевної порожнини – без особливостей. Огляд офтальмолога: очні середовища, дно – без особливостей, міопія 0І. Аудіограма: слух у нормі.
Лікування. Через 6 місяців із моменту встановлення діагнозу розпочато ФЗТ агалсидазою бета (препаратом Фабразим®*) в/в крапельно один раз на 2 тижні, симптоматичне лікування (знеболювальні, антигіпертензивні засоби). Упродовж 12-річної безперервної терапії агалсидазою бета отримано дуже сприятливий клінічний ефект: парестезії рідкісні, менш інтенсивні, не потребують приймання знеболювальних; не відзначаються підвищення АТ й температури тіла, збільшення кількості ангіокератом; спостерігаються поліпшення якості життя, активна соціальна діяльність, регулярні фізичні навантаження.
Не відзначено прогресування кардіоміопатії, натомість має місце стабільність серцевих проявів; толерантність до фізичних навантажень висока, діагностичних ознак порушення ритму та провідності не зафіксовано.
Пацієнт продовжує отримувати патогенетичну (ФЗТ) та симптоматичну терапію (іАПФ, вітамін D, карбамазепін ситуаційно).
Клінічний випадок 2
Пацієнт М., 24 роки.
Скарги. Біль у кінцівках, непереносимість спеки, висипання на долонях, зниження потовиділення, епізоди підвищення АТ і температури тіла, зниження якості життя.
Анамнез. У віці 7 років мали місце гастроінтестинальні розлади, часті бактеріальні інфекції, у 10 років – неприємні відчуття в дистальних відділах кінцівок з іррадіацією у проксимальні відділи, зі збільшенням інтенсивності при фізичних навантаженнях, кліматичних змінах, супутніх інфекціях із підйомом температури тіла до фебрильних цифр; протеїнурія, висипання на долонях. Діагноз: поліартрит неясної етіології.
У 23 роки: перший напад (ТІА) з парезом лівої частини обличчя та самостійним відновленням функцій м’язів за тиждень. У 24 роки: ішемічний інсульт у правій гемісфері, спастичний геміпарез зліва, парез мімічних м’язів за центральним типом зліва; госпіталізований із підозрою на ХФ.
Анамнез життя: без особливостей. Генеалогічний анамнез: молодший брат не хворіє на ХФ та не має клінічних проявів; мати з тяжкими серцевими проявами (гіпертрофічна кардіоміопатія) та болем у долонях і стопах, ТІА в анамнезі у віці 25‑30 років, її брат помер у 40 років від хронічної хвороби нирок, а бабуся пацієнта – у 40 років від інсульту.
Під час огляду: грубі риси обличчя, астенічна будова тіла, суха шкіра, гіпогідроз, поодинокі ангіокератоми на долонях.
Обстеження. Мікроальбумінурія – 1113 мг/добу (норма <30 мг), ЕКГ: синусова дихальна аритмія. ЕхоКГ: ТЗСЛШ – 10 мм (норма 6‑9), ТМШП – 10 мм (норма 6‑9). Активність α-галактозидази в лейкоцитах крові – 7,8 мкм/л/год (референтні значення – 43‑67 мкм/л/год), молекулярно-генетичне обстеження методом прямого автоматичного секвенування виявило мутацію в гені GLA у гемізиготному стані, клас патогенності перший. ХФ підтверджено.
Лікування. Призначено ФЗТ препаратом Фабразим®*, симптоматичну терапію (іАПФ, карбамазепін, антиагреганти), мультидисциплінарний підхід.
* Лікарський засіб Фабразим®, порошок для приготування концентрату 5 мг/мл для розчину для інфузій, зареєстрований в Україні. Р.П. № UA/10306/01/01. Наказ МОЗ України № 673 від 18.03.2020.
Важливість ранньої діагностики
Наведені клінічні випадки ілюструють важливість ранньої діагностики та своєчасного початку ФЗТ до появи незворотних загрозливих життю змін. Отже, у практичній роботі з пацієнтами, що страждають на ХФ, однією з найсерйозніших проблем залишається раннє виявлення захворювання.
Мультисистемний характер, клінічний поліморфізм, прогредієнтний перебіг, а також відсутність настороженості лікарів щодо можливого орфанного захворювання в кожному конкретному випадку ускладнюють своєчасну діагностику. Підвищення інформованості клініцистів будь-якого фаху сприятиме своєчасному встановленню діагнозу на ранньому етапі хвороби, коли патогенетична терапія є найефективнішою, що надасть можливість збільшити тривалість та підвищити якість життя хворих.
Своєчасна діагностика ХФ і призначення патогенетичної терапії виключає прояви інвалідизувальних симптомів та може сприяти повній реабілітації хворих із цією тяжкою недугою.
Ферментозамісна терапія хвороби Фабрі: нові дані щодо ефективності різних дозувань
Мета дослідження
Оцінити клінічну стабільність та безпеку після переведення пацієнтів з агалсидази бета в дозуванні 11 мг/кг/2 тижні на агалсидазу альфа по 0,2 мг/кг/2 тижні, а також результати після зворотного переходу на агалсидазу бета в дозуванні 1 мг/кг/2 тижні.
Дизайн дослідження
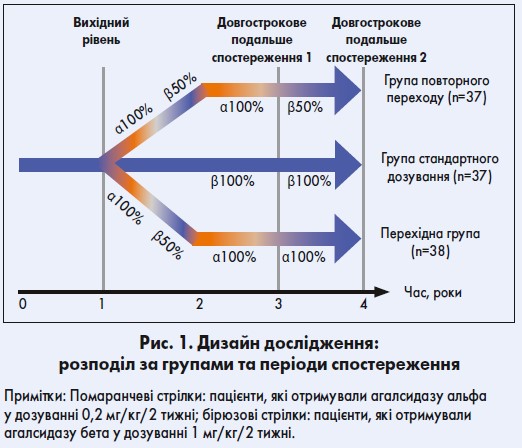 У процесі проспективного обсерваційного дослідження 112 пацієнтів (43 з яких були жіночої статі), що були стабільними на агалсидазі бета 1 мг/кг/2 тижні протягом щонайменше 12 місяців, у нерандомізованому порядку розподілили на три групи; спостереження проводили протягом 53 (діапазон – 38‑57) місяців (рис. 1):
У процесі проспективного обсерваційного дослідження 112 пацієнтів (43 з яких були жіночої статі), що були стабільними на агалсидазі бета 1 мг/кг/2 тижні протягом щонайменше 12 місяців, у нерандомізованому порядку розподілили на три групи; спостереження проводили протягом 53 (діапазон – 38‑57) місяців (рис. 1):
- група стандартного дозування: 37 хворих продовжували отримувати агалсидазу бета у дозуванні 1 мг/кг/2 тижні;
- перехідна група: 38 пацієнтів отримували знижену дозу агалсидази бета і згодом переходили на агалсидазу альфа в дозуванні 0,2 мг/кг/2 тижні або одразу ж – на агалсидазу альфа по 0,2 мг/кг/2 тижні і лишалися на агалсидазі альфа 0,2 мг/кг/2 тижні;
- група повторного переходу: 37 хворих знову переводили на агалсидазу бета у дозуванні 1 мг/кг/2 тижні після того, як вони отримували агалсидазу альфа по 0,2 мг/кг/2 тижні протягом щонайменше 12 місяців.
Середня тривалість терапії агалсидазою бета у дозуванні 1 мг/кг/2 тижні до включення пацієнта в дослідження становила в середньому 31 (12‑60) місяць. Дані хворого за попередній період використовували як контрольні, що давало можливість проаналізувати індивідуальні наслідки зміни лікування.
Вихідні характеристики
Пацієнти, які продовжували отримувати агалсидазу бета у дозуванні 1 мг/кг/2 тижні протягом усього дослідження, мали тяжчі ураження, ніж у перехідній групі та групі повторного переходу:
- достовірно більше хворих чоловічої статі, у яких була нижча активність α-галактозидази A (GLA) порівняно з іншими групами (обидва р<0,05);
- частіші симптоми, пов’язані з хворобою Фабрі (такі як ангіокератоми, діарея, гіпогідроз та напади болю, асоційованого з хворобою Фабрі), порівняно з пацієнтами перехідної групи (р<0,05);
- більша кількість балів за шкалою Майнца для оцінки ступеня тяжкості захворювання (MSSI) порівняно з пацієнтами групи повторного переходу (р<0,05).
Ключові результати
Функція нирок
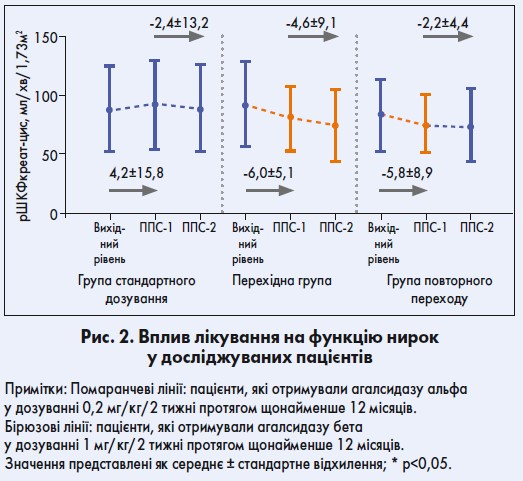 У пацієнтів групи повторного переходу на агалсидазу бета в дозуванні 1 мг/кг/ 2 тижні спостерігалося зменшення значень рШКФ (р<0,05). Щорічне зниження показників рШКФ від періоду подальшого спостереження 1 (ППС‑1) до періоду подальшого спостереження 2 (ППС‑2) становило -4,6 мл/хв/1,73 м2 у перехідній групі порівняно з -2,2 мл/хв/1,73 м2 у групі повторного переходу. У хворих, які отримували стандартне дозування, рівень рШКФ лишався стабільним протягом усього періоду подальшого спостереження (рис. 2).
У пацієнтів групи повторного переходу на агалсидазу бета в дозуванні 1 мг/кг/ 2 тижні спостерігалося зменшення значень рШКФ (р<0,05). Щорічне зниження показників рШКФ від періоду подальшого спостереження 1 (ППС‑1) до періоду подальшого спостереження 2 (ППС‑2) становило -4,6 мл/хв/1,73 м2 у перехідній групі порівняно з -2,2 мл/хв/1,73 м2 у групі повторного переходу. У хворих, які отримували стандартне дозування, рівень рШКФ лишався стабільним протягом усього періоду подальшого спостереження (рис. 2).
Гастроінтестинальні симптоми
Повторний перехід на агалсидазу бета у дозуванні 1 мг/кг/2 тижні приводив до значного зниження частоти випадків діареї (р<0,05). У пацієнтів перехідної групи частіше повідомлялося про діарею під час першого довгострокового періоду подальшого спостереження (р<0,05). У хворих, які отримували регулярні дози, симптоми з боку шлунково-кишкового тракту залишалися стабільними протягом усього періоду подальшого спостереження.
Lyso-GL‑3
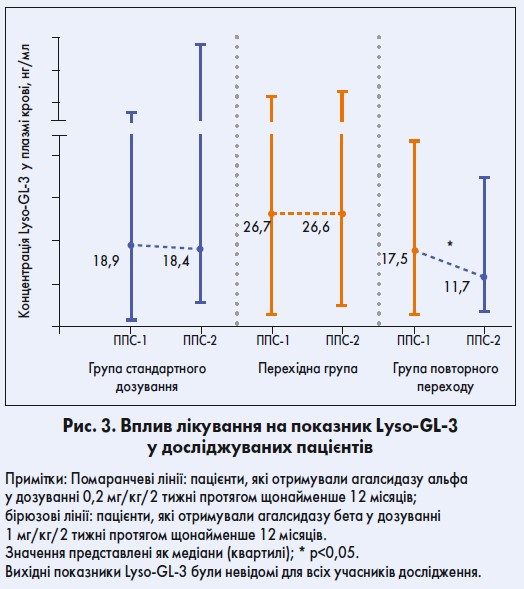 У групі повторного переходу на агалсидазу бета 1 мг/кг/2 тижні спостерігалося значне зниження рівнів глоботріаозилсфінгозину (Lyso-GL‑3); р<0,05 (рис. 3). Рівні Lyso-GL‑3 лишалися стабільними між ППС‑1 і ППС‑2 у групі стандартного дозування та перехідній. У пацієнтів перехідної групи відзначалися найвищі значення Lyso-GL‑3 під час ППС‑1 (р<0,05, порівняно з групами стандартного дозування і повторного переходу).
У групі повторного переходу на агалсидазу бета 1 мг/кг/2 тижні спостерігалося значне зниження рівнів глоботріаозилсфінгозину (Lyso-GL‑3); р<0,05 (рис. 3). Рівні Lyso-GL‑3 лишалися стабільними між ППС‑1 і ППС‑2 у групі стандартного дозування та перехідній. У пацієнтів перехідної групи відзначалися найвищі значення Lyso-GL‑3 під час ППС‑1 (р<0,05, порівняно з групами стандартного дозування і повторного переходу).
Безпека
Повторний перехід на агалсидазу бета у дозуванні 1 мг/кг/2 тижні добре переносився і не призводив до клінічно значущих інфузійних побічних реакцій. У трьох (8%) пацієнтів групи повторного переходу спостерігалися незначні інфузійні реакції (несуттєве підвищення температури тіла і надмірна стомлюваність).
Висновки
Повторний перехід на агалсидазу бета у дозуванні 1 мг/кг/2 тижні мав позитивний вплив на функцію нирок, симптоми з боку шлунково-кишкового тракту та рівні Lyso-GL‑3. У пацієнтів, які були повторно переведені на агалсидазу бета в дозуванні
1 мг/кг/2 тижні, спостерігалися:
- уповільнення погіршення функції нирок;
- статистично значуще зниження частоти випадків діареї;
- статистично значуще зменшення рівнів lyso-GL‑3 у плазмі крові.
Хворі перехідної групи мали постійне зниження рівнів рШКФ і частіше повідомляли про діарею. У пацієнтів, які отримували агалсидазу бета у дозуванні 1 мг/кг/2 тижні, частота випадків діареї, бали за MSSI та показники функції нирок залишалися стабільними.
Адаптовано за J. Krämer et al. Nephrol Dial Transplant, 2017: 1-11; doi: 10.1093/ndt/gfx319
Підготувала Олександра Демецька
Спецвипуск «Інсульт», Додаток до № 1 (56), 2021 р.
