


24 березня, 2021
Ювенільна системна склеродермія: діагностика, лікування, профілактика
«Системна склеродермія – загадка нашого покоління, драматична і несподівана при появі, унікальна і містична в своїх клінічних проявах, прогресуюча, яка наполегливо чинить опір лікуванню та призводить до відчаю і пацієнтів, і лікарів...»
Е. Bayoters, 1985
 Системна склеродермія (ССД) – гетерогенне системне захворювання сполучної тканини, для якого характерні васкулопатія малих судин, продукція аутоантитіл та дисфункція фібробластів із посиленим осадженням позаклітинного матриксу. Прогресуючий фіброз та розповсюджена судинна патологія за типом облітеруючої мікроангіопатії призводить до розвитку генералізованого синдрому Рейно (СР), індуративних змін шкіри, уражень опорно-рухового апарату та внутрішніх органів (легень, серця, нирок, шлунково-кишкового тракту).
Системна склеродермія (ССД) – гетерогенне системне захворювання сполучної тканини, для якого характерні васкулопатія малих судин, продукція аутоантитіл та дисфункція фібробластів із посиленим осадженням позаклітинного матриксу. Прогресуючий фіброз та розповсюджена судинна патологія за типом облітеруючої мікроангіопатії призводить до розвитку генералізованого синдрому Рейно (СР), індуративних змін шкіри, уражень опорно-рухового апарату та внутрішніх органів (легень, серця, нирок, шлунково-кишкового тракту).
Епідеміологія. Захворюваність на ССД серед дорослих складає 0,45-1,4 випадка на 100 тис. населення в рік, а у дітей – 0,05 випадка на 100 тис. Менше ніж у 10% усіх пацієнтів системний склероз розвивається у віці до 20 років; частка дітей до 16 років серед загальної кількості хворих становить менше 3%, а до 10 років – 2%. Середній вік дебюту захворювання у дітей – 8,1 року, найменший вік початку системного склерозу зареєстровано у дитини першого року життя. Дівчата до 16 років хворіють частіше, ніж хлопчики, у співвідношенні 3,6:1, представники негроїдної раси – частіше, ніж європеоїдної.
Етіологія. Припускають мультифакторний генез ССД, який складається зі спадкової схильності, впливу несприятливих екзо- та ендогенних чинників: інфекційних агентів, хімічних токсичних речовин, окремих лікарських препаратів, стресу, охолодження, інсоляції, оперативних втручань, фізичної травми, вакцинації, ендокринних зсувів тощо.
Генетична детермінованість хвороби підтверджується підвищеною частотою її виявлення у родичів пробандів і монозиготних близнюків; наявністю хромосомних аномалій у 95% хворих на ССД, виявленням у хворих генів гістосумісності HLA системи A9, В8, В35, DR1, DR3, DR5, DR11, DR52 та С4А. Встановлені кореляційні зв’язки між імуногенетичними маркерами, наявністю специфічних аутоантитіл і клінічними варіантами ССД. Так, антицентромерні антитіла поєднані з НLА-DR1, НLА-DR4, лімітованим ураженням шкіри, легеневою гіпертензією і хронічним перебігом, а анти-scl-70-антитіла – із НLА DRЗ, DR5, DQ7, дифузним ураженням шкіри, фіброзом легень і швидкопрогресуючим перебігом ССД.
Інфекційними тригерними факторами вважаються групи ретро-, герпесвірусів із характерною для них схильністю до персистенції та наявністю латентних і ендогенних форм. Згідно з вірусно-генетичною гіпотезою, трансплацентарна передача латентних вірусів від матері дитині може стимулювати спадкову природу захворювання.
Патогенез. Провідну роль у патогенезі відіграють 3 компоненти: імунна активація, ушкодження ендотелію (судин), надмірний синтез позаклітинного матриксу з посиленим відкладенням структурно нормального колагену. Підвищення колагено- та фіброутворення посідає центральне місце у патогенезі й визначає нозологічну специфіку захворювання. Серед порушень у Т- і В-ланках імунітету відзначають: Т-клітинну активацію та дисрегуляцію Т-хелперів (Th1 і Th2), дисбаланс імунорегуляторних субпопуляцій зі зниженням кількості Т-супресорів, сенсибілізацію лімфоцитів крові людини до антигенів шкіри, рівень яких корелює зі ступенем зниження Т-супресорів та стадією склеродермії (рис. 1). Гіперпродукція активованими Т-лімфоцитами цитокінів (ІЛ-1, 4, 6) стимулює проліферацію В-лімфоцитів із посиленням синтезу специфічних аутоантитіл. Активовані клітини імунної системи, ендотеліальні клітини і фібробласти здатні продукувати цитокіни та фактори росту, які можуть чинити паракринний або аутокринний вплив на інші клітини. Прозапальні цитокіни активують проліферацію фібробластів і пригнічують їх апоптоз.
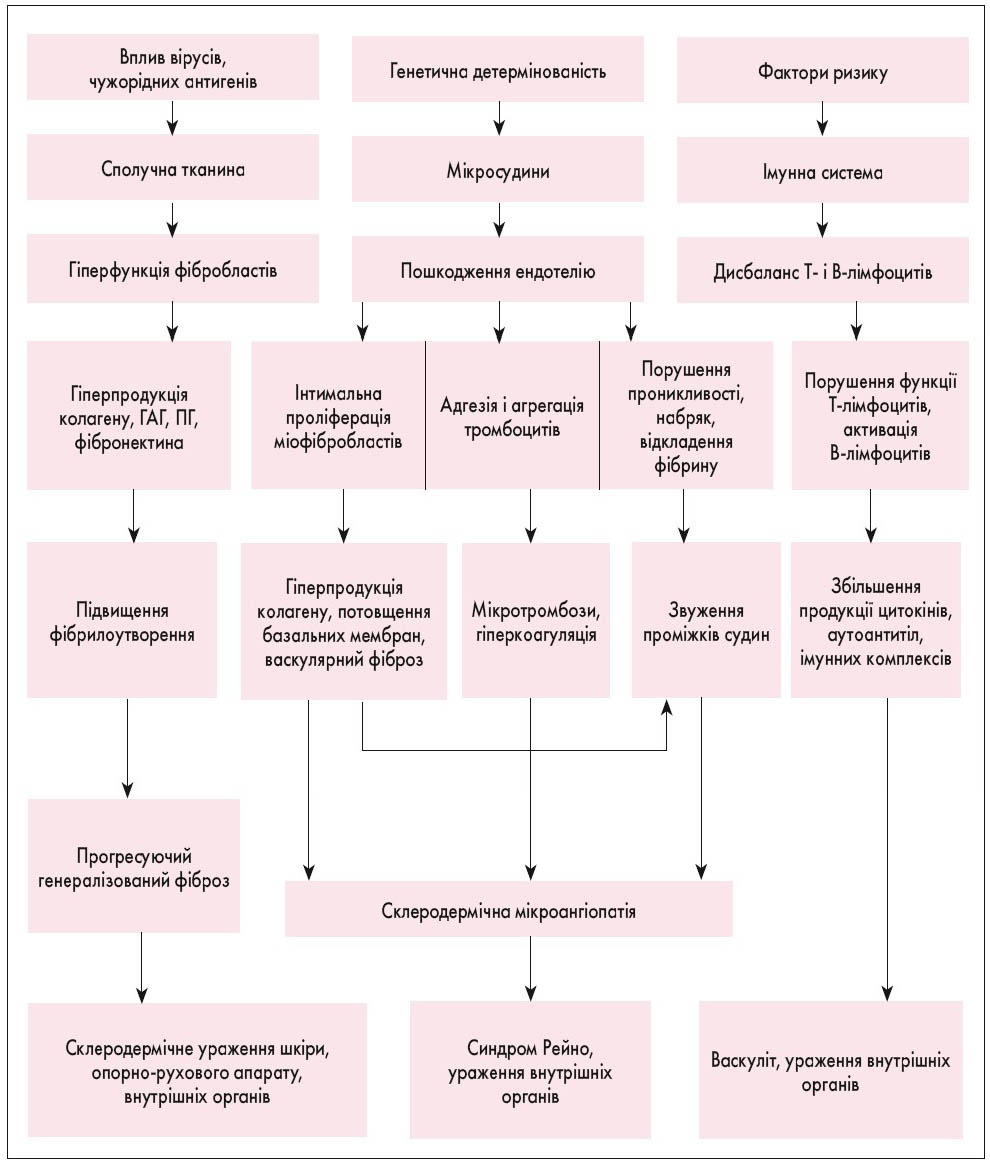 Рис. 1. Схема патогенезу склеродермії (В.І. Мазуров, 2005)
Рис. 1. Схема патогенезу склеродермії (В.І. Мазуров, 2005)
Інтенсивна синтетична активність фібробластів проявляється посиленням синтезу колагену I, III типів і компонентів міжклітинного матриксу, що призводить до розвитку регіонального або генералізованого фіброзу (рис. 2). Цитотоксичні лімфоцити ушкоджують клітини ендотелію, що призводить до адгезії і агрегації тромбоцитів, активації процесів згортання крові, вивільнення медіаторів запалення, збільшення проникливості стінок судин із периваскулярною запальною інфільтрацією.
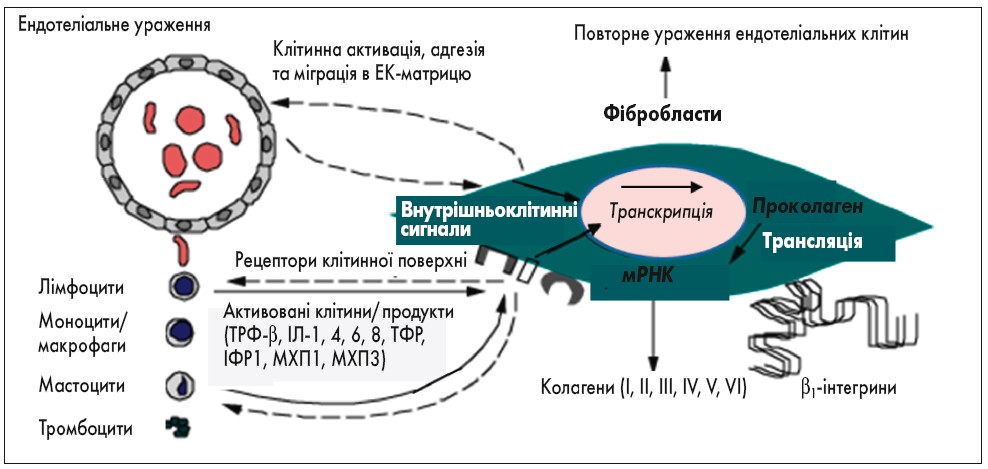 Рис. 2. Паракринні міжклітинні взаємодії при склеродермії: ТРФ-β – трансформуючий ростовий фактор β, ІЛ-1, 4, 6, 8 – інтерлейкіни, ТФР – тромбоцитарний фактор росту, ІФР-1 – інсуліноподібний фактор росту 1, МХП-1, 3 – хемотаксичні білки моноцитів 1, 3 (P. Christopher, 2018)
Рис. 2. Паракринні міжклітинні взаємодії при склеродермії: ТРФ-β – трансформуючий ростовий фактор β, ІЛ-1, 4, 6, 8 – інтерлейкіни, ТФР – тромбоцитарний фактор росту, ІФР-1 – інсуліноподібний фактор росту 1, МХП-1, 3 – хемотаксичні білки моноцитів 1, 3 (P. Christopher, 2018)
Порушення мікроциркуляції супроводжується проліферацією гладком’язових клітин, гіперплазією інтими та звуженням діаметру судин, деформацією капілярної сітки, стазом крові. Дефіцит судинорозширювальних нейропептидів, вазоактивних субстанцій (оксиду азоту, ендотеліну-1), активація тромбоцитів зі збільшенням виділення тромбоцитарного фактора росту та наступною проліферацією фібробластів і синтезом колагену посилюють судинний спазм, що призводить до розвитку СР, легеневої та реноваскулярної гіпертензії. На тлі запального ураження стінки судин, вазоспазму змінюються внутрішньосудинні плазмові та клітинні властивості крові: підвищується в’язкість крові з тенденцією до гіперкоагуляції, пригнічується фібриноліз та агрегація формених елементів. Унаслідок порушень мікроциркуляції, процесів гемостазу та судинного спазму виникають ішемічно-некротичні ураження в органах і тканинах.
Патоморфологія ССД характеризується дифузним ураженням сполучної тканини у вигляді мукоїдного, фібриноїдного набухання, фібриноїдного некрозу, гіалінозу, склерозу, продуктивного васкуліту.
На ранніх стадіях хвороби у нижніх шарах дерми підвищується вміст Т-лімфоцитів, моноцитів, плазматичних і тучних клітин. На пізніх стадіях епідерміс стає тоншим, у дермі паралельно йому розташовуються потовщені гомогенні пучки колагену, які проростають із дерми у підшкірну клітковину, через що шкіра тісно спаюється із тканинами. Придатки шкіри атрофуються, стоншується епідерміс.
При уражені шлунково-кишкового тракту (ШКТ) фібротичні зміни виражені нечітко. Слизова середньої і нижньої третини стравоходу стоншена, у власній пластинці слизової і підслизовому шарі підвищений вміст колагену. Власна пластинка слизової стравоходу й інших відділів ШКТ інфільтрована лімфоцитами і плазматичними клітинами, ураження м’язової оболонки проявляється атрофією та фіброзним переродженням гладких м’язів. При атрофії м’язової оболонки товстої кишки утворюються дивертикули та відбувається дилатація окремих відділів ШКТ. Ураження легень проявляється дифузним інтерстиціальним і перибронхіальним фіброзом, посиленою проліферацією епітелію бронхів і потовщенням стінок альвеол. Розрив стінок альвеол може призводити до появи дрібних кіст і бульозної емфіземи легень.
У синовіальній оболонці хворих на ССД із ураженням суглобів визначаються набряк та інфільтрація лімфоцитами і плазматичними клітинами, відкладення фібрину, на пізніх стадіях має місце фіброз синовіальної оболонки. Міопатія гістологічно проявляється інтерстиціальною і периваскулярною лімфоцитарною інфільтрацією, дегенерацією м’язових волокон та інтерстиціальним фіброзом. Спостерігається потовщення стінок артеріол і зменшення кількості капілярів.
Процеси фіброзування і порушення мікроциркуляції розвиваються в усіх оболонках серця (міокарді, ендокарді й перикарді). У міокарді визначається дегенерація кардіоміоцитів та інтерстиціальний фіброз, найбільш виражений навколо судин. Можуть потовщуватися стінки дрібних коронарних артерій. У деяких хворих розвивається фібринозний перикардит із перикардіальним випотом та фіброз провідникової системи серця.
При гістологічному дослідженні нирок виявляється гіперплазія інтими міждолькових артерій, фібриноїдний некроз клубочків і артеріол, потовщення базальної мембрани клубочків.
Класифікація. Системну склеродермію за МКХ-10 відносять до класу ХIII «системні захворювання кістково-м’язової та сполучної тканини», M30-M36 «системні ураження сполучної тканини».
M34 системний склероз:
- M34.0 прогресуючий системний склероз;
- M34.1 синдром CREST;
- M34.2 системний склероз, спричинений медикаментозними засобами і хімічними сполученнями;
- M34.8 інші форми системного склерозу;
- M34.9 системний склероз неуточнений.
Класифікації ССД для дітей сьогодні не існує, тому в клінічній практиці застосовується робоча класифікація для дорослих (АРУ, 2004).
Клінічні форми:
- дифузна ССД (dcSSc):
- генералізоване ураження шкіри обличчя, тулуба, проксимальних і дистальних відділів кінцівок протягом року після виникнення СР;
- ранній (протягом першого року хвороби) розвиток вісцеральної патології (інтерстиціальне ураження легень, олігоурична ниркова недостатність, ураження міокарда і ШКТ);
- значне розширення капілярів ложа нігтів із формуванням аваскулярних вогнищ (за даними капіляроскопії);
- виявлення антитіл до топоізомерази-1 (анти-Scl-70);
- лімітована (акросклеротична) ССД (lcSSc):
- тривалий період ізольованого СР, який передує змінам шкіри;
- ураження шкіри обмежене (дистальні відділи передпліччя і кистей, гомілки, стопи);
- розвиток CREST-синдрому (С – кальциноз, R – синдром Рейно, Е – езофагіт, S – склеродактилія, Т – телеангіоектазія);
- розширення капілярів ложа нігтів без аваскулярних вогнищ (за даними капіляроскопії);
- виявлення антицентромірних антитіл (АЦА);
- перехресна ССД (overlap-syndrome) – поєднання ознак ССД із іншими системними захворюваннями сполучної тканини (дермато/поліміозит, ревматоїдний артрит, системний червоний вовчак та ін.): ССД-РА, ССД-ДМ/ПМ, ССД-СЧВ;
- ювенільна ССД (ЮССД) характеризується початком захворювання до 16 років та має особливості клінічного перебігу;
- вісцеральна ССД – переважає ураження внутрішніх органів і СР, шкірні зміни мінімальні або відсутні. Ця форма вважається відносно рідкою, можливо, у зв’язку із труднощами діагностики та правильного трактування захворювання;
- індукована СД: розповсюджене частіше дифузне ураження шкіри (індурація) у поєднанні із судинною патологією, яке розвинулося після впливу хімічних або інших факторів навколишнього середовища;
- пресклеродермія: клінічно ізольований СР у поєднанні з капіляроскопічними змінами за наявності позитивних антиядерних антитіл (ANA) та специфічних аутоантитіл (Scl-70 та антицентромірних). Діти зі пресклеродермією потребують ретельного моніторингу, оскільки у них доведено вищий ризик прогресування із розвитком системного склерозу.
Варіанти перебігу:
- гострий (злоякісний), швидко прогресуючий (генералізований фіброз): дифузний фіброз шкіри, ураження внутрішніх органів (фіброзні зміни легень), судинна патологія (склеродермічна нирка) у перші 2 роки від початку захворювання, висока летальність;
- підгострий, помірно прогресуючий: переважають ознаки аутоімунного запалення (щільний набряк шкіри, артрит, міозит), нерідко мають місце ознаки overlap-синдрому;
- хронічний, повільно прогресуючий: переважає судинна патологія – на початку захворювання тривалий СР із поступовим розвитком помірних шкірних змін (лімітована форма), наростання судинних ішемічних розладів, вісцеральної патології (ШКТ, нирок, серця, легенева гіпертензія).
Прогноз двох останніх варіантів перебігу більш сприятливий.
Стадії:
- I (початкова) характеризується 1-3 локалізаціями (СР, суглобовий синдром, щільний набряк, інколи – вісцерит);
- II (генералізована) відображає системний, полісиндромний характер процесу: індурація шкіри, контрактури, полівісцеральна патологія (легені, серце, ШКТ, нирки);
- III (термінальна) – розвивається недостатність хоча б одного життєво важливого органу, залученого до патологічного процесу (легень, серця, нирок).
Приклад формулювання клінічного діагнозу. Ювенільна системна склеродермія, дифузна форма, гострий перебіг, II стадія. Щільний набряк/індурація шкіри (локалізація). Синдром Рейно. Ураження стравоходу (гіпотонія), початковий інтерстиціальний фіброз легень.
Клінічна картина. Для склеродермії характерні ураження шкіри, опорно-рухового апарату, судин у поєднанні з вісцеральною патологією.
Вазомоторні порушення у вигляді спастичних кризів – СР спостерігаються у 75% дітей, може з’явитися задовго до інших симптомів. Тривалий час симптоми можуть бути стертими або виникати під впливом холоду, стресу. На початку нападу ділянка шкіри має бліде забарвлення, а через декілька хвилин стає синьо-фіолетового відтінку, пацієнт відчуває холод, оніміння, порушується чутливість. Після закінчення спазму шкіра стає рожевою або червоною, пацієнт відчуває біль (табл. 1). 
У період між нападами колір шкіри на пальцях ніг, язиці, кінчику носа, вушних раковинах і підборідді може бути цианотичним, зберігаються парестезії і больовий синдром. При тривалому перебігу СР розвиваються ускладнення: трофічні зміни шкіри пальців, вушних раковин, повік, виразки шкіри над кістками, некроз і гангрена пальців. Порушення трофіки і гіпоксія тканин лежать в основі остеолізу дистальних фаланг пальців.
Генералізовані судинні кризи із втягненням у патологічний процес судин головного мозку і внутрішніх органів у вигляді вазоспастичних порушень у серці, легенях, нирках зустрічаються не часто.
Ураження шкіри у дітей локалізуються на будь-якій ділянці тіла: тулубі, обличчі, кінцівках, сідницях. Типові склеродермічні зміни: щільний набряк, індурація, гіпер- або депігментація, атрофія, алопеція, кальциноз.
Виділяють 3 стадії ураження шкіри.
1. Набряк шкіри щільний, безболісний, шкіра не збирається у складки. Наявні вогнища судинного стазу, плями з ліловим вінчиком по периферії, колір шкіри – від білого до синюшно-рожевого.
Виразність ущільнення шкіри оцінюється методом пальпації за 4-бальною шкалою: 0 – ущільнення немає; 1 – ущільнення незначне; 2 – ущільнення помірне; 3 – ущільнення значне (шкіра не збирається у складку).
2. Стадія індурації – потовщення шкіри тістоподібної консистенції. Шкіра біло-жовта, воскоподібна, щільно спаяна з прилеглими тканинами, блискуча, із ділянками гіпер- і депігментації, на тулубі – множинні телеангіектазії на «бронзовому» тлі, синдром брудної шкіри. Обличчя стає маскоподібним, із потовщеною шкірою чола, щік, витонченими губами, глибокі зморшки біля рота (симптом кисету), спинка носа – витончена, крила – напружені (так званий дзьоб хижака). Шкірні зміни часто поєднуються із судинною і трофічною патологією: виразками, гнійниками, трофічними змінами нігтів, алопецією.
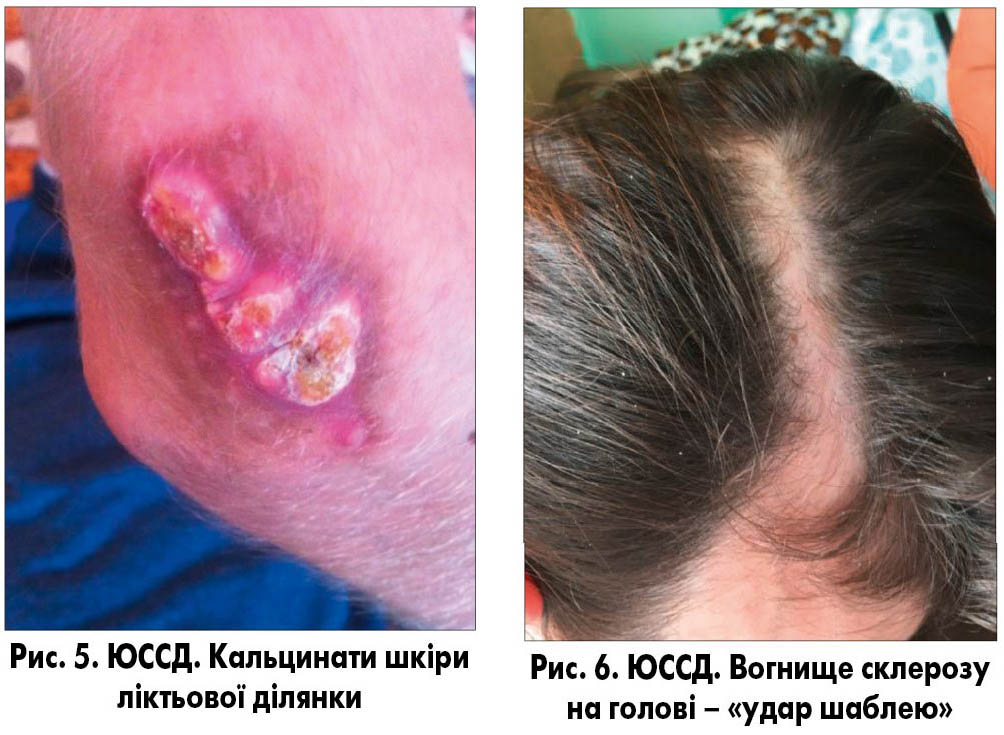
3. Стадія склерозу і атрофії: шкіра щільна, потовщена, має характерний блиск, жовтувате забарвлення. Порушується робота сальних, потових залоз, розвивається атрофія волосяних фолікулів (рис. 3). При тривалому перебігу хвороби можливе відкладання солей кальцію (кальцинатів), які виявляються при рентгенологічному або ультразвуковому обстеженні (рис. 4). Специфічним симптомом є поява кальцинатів у підшкірній клітковині пальців рук, частіше на нігтьових фалангах. Може розвинутися кальциноз надгортанника, голосових зв’язок, перикарда, міокарда, клапанів серця, капсули печінки і селезінки.
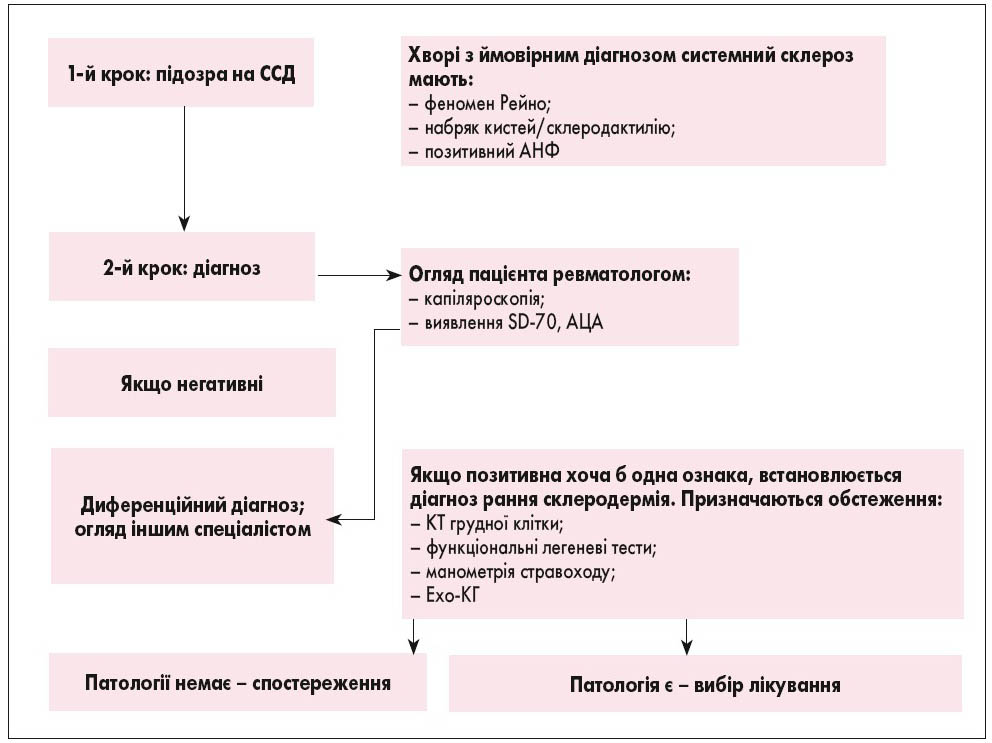 Рис. 3. Алгоритм діагностики ранньої склеродермії (M. Matucci-Cerinic et al., 2009)
Рис. 3. Алгоритм діагностики ранньої склеродермії (M. Matucci-Cerinic et al., 2009)
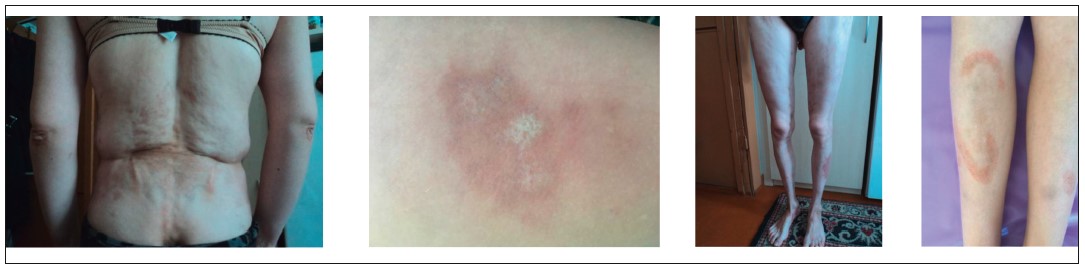 Рис. 4. ЮССД. Типові склеродермічні ураження шкіри: набряк, індурація, гіпер-, депігментація, атрофія
Рис. 4. ЮССД. Типові склеродермічні ураження шкіри: набряк, індурація, гіпер-, депігментація, атрофія
Залежно від розповсюдженості і характеру уражень шкіри, виділяють декілька варіантів шкірного синдрому у дітей:
- дифузне швидке, тотальне, індуративне ураження шкіри (синдром корсета або панциря), що обмежує екскурсію грудної клітки;
- лімітоване (акросклеротичне) ураження дистальних відділів кінцівок (кистей рук, стоп); пальці стають набряклими, сосископодібними, не стискаються в кулак (склеродактилія), формуються контрактури, кисть стає як кігтиста лапа; характерний СР із порушенням трофіки кінцевих фаланг пальців рук і ніг, розвитком дигітальних рубців, пренекрозів; досить часто спостерігаються телеангіектазії (локальні розширення капілярів і дрібних судин);
- проксимальне ураження кінцівок вище зап’ясткових і плесневих суглобів;
- гемісклеродермія – ураження однієї кінцівки й однобічне ураження тулуба і кінцівок, інколи із розповсюдженням вогнища на шию, частину обличчя; глибокі трофічні зміни на цих ділянках призводять до зменшення об’єму кінцівок і порушень їх росту;
- атипова – стерте або вогнищеве ураження шкіри.
Ураження слизових оболонок (хронічний кон’юнктивіт, кератит, фарингіт, атрофічний риніт, стоматит) у дітей спостерігається не часто.
Ураження суглобів і кісток – один із найбільш частих (50-70%) клінічних проявів у дітей, а в 30% випадків він діагностується як перший симптом захворювання. Виділяють 3 варіанти суглобового синдрому: поліартралгія, поліартрит, поліартрит із розвитком контрактур.
Поліартралгія спостерігається зранку, в основі лежать ураження структур зв’язкового апарату, шкіри з переродженням колагено-еластинової основи на фіброзну та ішемізацією навколосуглобових ділянок. Поліартрити з переважно ексудативними або фіброзно-індуративними змінами призводять до деформації суглобів за рахунок контрактур без рентгенологічних ознак ураження саме суглобів. Ерозія суглобових поверхонь та епіфізарний остеопороз спостерігаються не часто.
Судинно-трофічні порушення та зміни у колагеновій матриці призводять до акроостеолізу дистальних фаланг, інколи відростків променевих кісток і нижньої щелепи.
При локалізації вогнища склерозу на обличчі та голові по типу «удар шаблею» формуються виражені деформації кісток черепу із западанням та стоншенням кісток, геміатрофією зубощелепного апарату (рис. 5-8).
 Ураження скелетних м’язів, сухожилля. При ССД розвивається фіброзуючий інтерстиціальний міозит із розростанням сполучної тканини й атрофією м’язів. Хворих турбують поліміалгія, різка м’язова слабкість, обмеження рухів. М’язи щільні, ригідні, м’язово-сухожильні контрактури з часом призводять до анкілозів суглобів.
Ураження скелетних м’язів, сухожилля. При ССД розвивається фіброзуючий інтерстиціальний міозит із розростанням сполучної тканини й атрофією м’язів. Хворих турбують поліміалгія, різка м’язова слабкість, обмеження рухів. М’язи щільні, ригідні, м’язово-сухожильні контрактури з часом призводять до анкілозів суглобів.
Ураження травного тракту можливо в усіх його відділах, однак найчастіше спостерігається ураження стравоходу і кишечнику (від 30 до 74%).
Склеродермічний езофагіт проявляється гіпотонією стравоходу з ознаками дисфагії (утруднення ковтання, зригування, блювання, розвиток рефлюкс-езофагіту). На слизовій оболонці стравоходу внаслідок дії кислого вмісту шлунка виникають подразнення, ерозія, виразки. Переродження гладкої мускулатури стравоходу у фіброзну тканину призводить до стриктур у нижній третині та розширення відділу над звуженням, що робить неможливим ковтання твердої їжі.
Залучення кишечнику до патологічного процесу маніфестує порушенням моторики (запор, діарея, біль) та процесів всмоктування, розвивається некроз, трофічні виразки стінки кишки із подальшою їх перфорацією.
Ураження легень спостерігається у 41-53% дітей у вигляді інтерстиціального базального фіброзу, фіброзуючого альвеоліту із формуванням кіст та поступовим розвитком легеневої гіпертензії. Фіброзний процес у плеврі може призводити до адгезивного плевриту, зрощення синусів, звуження плевральної порожнини.
Клінічно при розвитку фіброзу легень виявляється задишка, кашель, аускультативно вислуховується розповсюджена двобічна крепітація у базальних відділах. При аускультації серця – акцент та роздвоєння другого тону над легеневою артерією та над трьохстулковим клапаном.
Інколи клінічні ознаки легеневого процесу відсутні. Зміни у легенях виявляються лише при інструментальних дослідженнях (рентгенографії, спірографії, КТ органів грудної клітки).
У 25-40% хворих розвиваються ураження серця по типу склеродермічного кардіосклерозу, який клінічно проявляється зміною скоротливої активності зі зниженням фракції викиду, порушеннями ритму та провідності. Зміни з боку ендокарда у вигляді фібропластичного ендокардиту із формуванням у подальшому клапанного склерозу у дітей зустрічається не часто. Можливим є розвиток фібринозного перикардиту. Ураження серця у дітей із ССД є основною причиною синдрому раптової смерті.
Нирки при ССД залучаються до патологічного процесу у 10-14% дітей. Розвивається склеродермічне ураження судин із формуванням хронічної склеродермічної нефропатії. У хворих спостерігається протеїнурія, гематурія, артеріальна гіпертензія з можливим формуванням ниркової недостатності. Істинна «склеродермічна» нирка (склеродермічний нирковий криз) клінічно проявляється значною протеїнурією, злоякісною гіпертонією, швидко прогресуючою нирковою недостатністю. 
Неврологічна симптоматика характеризується розвитком поліневротичного синдрому, прогресуючих тригемінальних сенсорних невропатій.
Ураження ендокринної системи виникає у вигляді тиреоїдиту зі зменшенням функції щитоподібної залози (тиреоїдит Хасимото). Можливе формування гіпер-, гіпофункції наднирників, розвиток цукрового діабету.
Особливості клінічного перебігу ЮCCД:
- переважають ураження шкіри по типу вогнищевої або лінійної (геміформи) склеродермії;
- СР стертий або помірно виражений;
- більш частий суглобовий синдром із розвитком стійких контрактур та м’язових уражень;
- скудна вісцеральна патологія, переважає функціональна недостатність органів, зменшена частота розвитку ренальних кризів та кардіопульмональних уражень;
- частий розвиток overlap-синдрому;
- помірна активність аутоімунного процесу;
- хронічний перебіг захворювання зі стійкими періодами ремісії.
Продовження в наступному номері.
Тематичний номер «Педіатрія» № 1 (57) 2021 р.
