


3 липня, 2021
Доброякісні гіпербілірубінемії (спадкові пігментні гепатози) та їхнє місце в практиці сімейного лікаря
 Для сучасного популяційного здоров’я населення притаманно погіршення структурно-функціонального стану печінки, спричинене численними екзогенними (синтетичні продукти харчування, алкоголь, лікарські засоби, хімічні речовини в побуті та на виробництві, гепатотропні віруси) й ендогенними (хвороби всіх систем, ожиріння) факторами. Ураження печінки належать до найпоширеніших у популяції, часто стають коморбідним тлом і значною мірою зумовлюють ефективність лікування та профілактики всіх інших хвороб, адже зміни функції печінки впливають на метаболізм медикаментів і подеколи обмежують можливості лікування [1].
Для сучасного популяційного здоров’я населення притаманно погіршення структурно-функціонального стану печінки, спричинене численними екзогенними (синтетичні продукти харчування, алкоголь, лікарські засоби, хімічні речовини в побуті та на виробництві, гепатотропні віруси) й ендогенними (хвороби всіх систем, ожиріння) факторами. Ураження печінки належать до найпоширеніших у популяції, часто стають коморбідним тлом і значною мірою зумовлюють ефективність лікування та профілактики всіх інших хвороб, адже зміни функції печінки впливають на метаболізм медикаментів і подеколи обмежують можливості лікування [1].
Ураження печінки завжди розвиваються стадійно: стеатоз → гепатит → цироз → гепатоцелюлярна карцинома. Проте клінічні, лабораторні та інструментальні прояви на стадіях гепатозу часто неспецифічні та неяскраві, що призводить до гіподіагностики та відсутності профілактичної настороженості. Тож особливого значення набувають генетично зумовлені спадкові пігментні гепатози – доброякісні гіпербілірубінемії (ДГБ) – ураження печінки дистрофічного характеру, що розвиваються з генетично детермінованих ензимопатій із порушенням внутрішньопечінкового обміну білірубіну та проявляються порушеннями функцій печінки.
Наукові погляди на клінічне значення пігментних гепатозів не одностайні: з одного боку, вони вважаються підґрунтям і початком значніших уражень печінки, а з другого – нещодавно описаними антиоксидантними властивостями білірубіну. Виявилося, що синдром Жильбера (СЖ) асоціюється зі вдвічі нижчим ризиком смерті від вік-залежних хвороб унаслідок антиоксидантних властивостей білірубіну [2], зокрема білірубіну IXα. Хоча зниження ризику кардіоваскулярних хвороб у пацієнтів із СЖ показано в декількох дослідженнях, більшою мірою це стосується осіб із певним генотипом – (TA)7/(TA)7, на відміну від звичайного генотипу (TA)6/(TA)6 [3]. Однак питання клінічного значення ДГБ є складнішим, адже, співіснуючи з пацієнтом усе життя, вони можуть істотно впливати на шляхи трансформації всіх ліків і ксенобіотиків в організмі, змінювати природний перебіг інших хвороб внутрішніх органів і безпосередньо ініціювати тяжкі форми уражень печінки та жовчовивідних шляхів. Тому вчасна діагностика, профілактика та раціональне лікування є важливими аспектами сучасної медичної практики [4].
Роль печінки в обміні білірубіну достатньо вивчена. Білірубін – це продукт метаболізму гемопротеїнів, які утворюються з гемоглобіну еритроцитів після їх розпаду. За добу в людини розпадається приблизно 1% еритроцитів, які містять 6-8 г гемоглобіну. Оскільки з 1 г гемоглобіну утворюється 35 мг білірубіну, добова його продукція теоретично має бути 220-290 мг, але реально цей показник є дещо нижчим і становить 200-250 мг. Цей вільний білірубін не розчиняється в рідинах, тому для його транспорту потрібен носій – білок крові альбумін, 1 моль якого може зв’язати 2 моля білірубіну. За нормального вмісту білка сироватки крові це становить 70 мг пігменту на 100 мл плазми. Насправді білірубіну зв’язується менше, бо деякі екзогенні й ендогенні речовини конкурують із ним за альбумін (кофеїн, саліцилати, сульфаніламіди). Первісний білірубін називається вільним, або непрямим, оскільки безпосередньо не взаємодіє з діазореактивом Ерліха й дає позитивну реакцію лише після осадження білків плазми крові. Прямий зв’язаний білірубін, який виявляється у зв’язку з глюкуронідом і дає пряму реакцію з діазореактивом Ерліха, є нетоксичним і легко проходить крізь біологічні мембрани. Вміст загального білірубіну в крові дорослої людини становить від 8,5 до 20,5 мкмоль/л, із них близько 75% – це вільний непрямий білірубін (6,5-15,4 мкмоль/л), а 25% – зв’язана фракція (2,1-5,1 мкмоль/л). Отже, в печінці відбуваються захоплення комплексу білірубін + альбумін із плазми, поєднання його з глюкуроновою кислотою й екскреція утвореної сполуки в жовчний капіляр. Порушення кожного етапу окремо чи декількох одночасно, зумовлене генетично, і спричиняє ДГБ.
Історія
Історія вивчення ДГБ налічує 120 років. Уперше синдром описав французький професор клінічної медицини Ніколя Огюстен Жильбер (Gilbert A., Lereboullet P., 1901), який багато зробив для розвитку гепатології. Тривалий час ДГБ (холемії, за термінологією Жильбера) не відокремлювали від хронічного гепатиту та гемолітичної анемії в самостійну нозологічну форму.

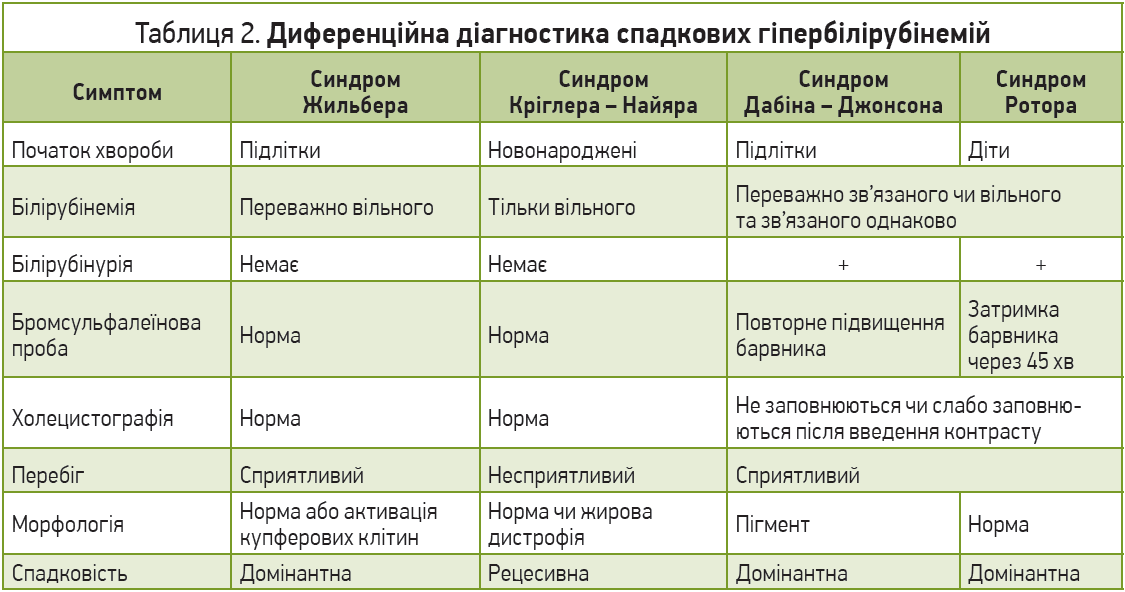
До 1961 року в СРСР було описано лише 30 таких клінічних спостережень (перший – у 1928 році доктором Ліфшицем). На українських теренах ДГБ вперше описав професор Львівського медичного університету Юліан Децик у 1963 році. Лише через 50 років від першого опису СЖ були відокремлені й охарактеризовані інші форми ДГБ – жовтяниці Кріглера – Найяра (Crigler J. F., Najjar V. A., 1952), Ротора (Rotor A. B.M.L., Florentin A. 1948), Дабіна – Джонсона (Dubin I. N., Johnson F. B., 1954), Люсі – Дріскол (Arias I. et al., 1965).





Класифікація
До ДГБ належать некон’юговані (1) та кон’юговані (2) форми:
1а) синдром Жильбера;
1b) синдром Кріглера – Найяра;
1c) синдром Люсі – Дріскол;
2a) синдром Дабіна – Джонсона;
2b) синдром Ротора (кон’югована та некон’югована ДГБ).
Радянський гепатолог А. Ф. Блюгер (1926-2007) виокремлював іще післягепатитну гіпербілірубінемію, яка визнається не всіма вченими, оскільки, на їхню думку, перенесений гепатит у таких пацієнтів лише зумовлює клінічну маніфестацію наявного генетичного дефекту.
Синдром Жильбера належить до найчастіших форм ДГБ, спостерігається майже в 5% населення, дещо рідше в азіатів і значно частіше в африканців (36%), у 10 разів частіше в чоловіків, аніж у жінок. СЖ успадковується за автосомно-домінантним типом, спричиняється мутаціями гена UGT1A1, які зумовлюють зменшення активності глюкоронілтрансферази‑1А1 [5]. Патогенез СЖ включає декілька ланок, які можуть мати різну вираженість: посилений гемоліз еритроцитів зі вкороченням тривалості їхнього життя, зменшення захоплення гепатоцитами білірубіну з крові та зниження кон’югаційної функції печінки. Виникнення жовтяниці зумовлено найперше порушенням кон’югації та захоплення білірубіну гепатоцитами, а другорядну роль відіграють підвищений гемоліз і порушення екскреції білірубіну з печінкової клітини. Морфологічно спостерігаються жирова інфільтрація печінки та помірне відкладення пігменту. Гістоензимологічні дослідження біоптату печінки виявляють зростання активності лужної фосфатази (ЛФ) печінкових і купферових клітин.
Клініка СЖ є неспецифічною. Незважаючи на спадковий характер патології, перші її прояви з’являються зазвичай у підлітковому чи молодому віці, коли людина починає виконувати значні фізичні навантаження та підлягає впливу інших несприятливих факторів на печінку (вживання алкоголю), що зумовлює часте виявлення СЖ під час військової служби. До появи жовтяниці (коли вміст білірубіну крові ≥34 мкмоль/л) діагностика зазвичай утруднена. Провокують жовтяницю перевтома, фізичне навантаження, інфекції жовчних шляхів, уживання ліків та алкоголю. Н. О. Жильбер описав характерну клінічну тріаду: жовтяниця, ксантелазми повік і печінкові маски. Проте, за нашими спостереженнями, ксантелазми повік і печінкові маски є пізніми проявами синдрому. Часто пацієнтів турбують також болі в животі різного характеру, диспепсія, астеновегетативні прояви. Фізикальне обстеження виявляє певне збільшення печінки, переважно помірне, іноді може збільшуватися селезінка, якщо синдром перебігає за так званим спленомегалічним, несприятливішим варіантом, описаним Н. О. Жильбером дещо пізніше – в 1907 році. За нашими спостереженнями, серед 63 обстежених військових із ДГБ, які проходили стаціонарне лікування, у 3% симптоми з’явилися після гострого гепатиту А, у 2% – після гострої респіраторно-вірусної інфекції, у 2% – після значного фізичного навантаження, 1% осіб відзначали наявність СЖ із дитинства, тоді як у решти (92%) прояви ДГБ виникли без явного провокувального фактора та маскувалися скаргами, притаманними ураженням стравоходу, шлунка та 12-палої кишки [6]. Тобто в молодих осіб із гастроентерологічними скаргами має бути діагностична настороженість щодо ДГБ.
Стандартне додаткове лабораторно-інструментальне обстеження досить легко дає змогу запідозрити ураження печінки, але для діагностики власне ДГБ та його форми є недостатньо специфічним, тому діагноз СЖ, як і всіх ДГБ, встановлюється переважно після виключення інших хвороб печінки, передусім гепатитів. Зміни скринінгових показників крові є невираженими: загальний аналіз крові відповідає нормі або еритроцити та гемоглобін є дещо зниженими внаслідок гемолізу зі зниженням осмотичної резистентності еритроцитів. Біохімічні показники характеризуються періодичним збільшенням білірубіну (переважно непрямого) й уробіліну сечі, періодично також підвищуються в крові маркери цитолізу та холестазу (передусім аланінамінотрансфераза (АЛТ) та ЛФ) на тлі нормальних значень проб, які відображають стабільність колоїдів сироватки крові (реакція Таката-Ара, Вельтмана, тимолова проба). Приблизно через 5 років від початку маніфестації хвороби можуть виникати гіпоальбумінемія та гіпохолестеринемія. Біохімічні зсуви свідчать про те, що в основі СЖ лежить ураження гепатоцита з ознаками цитолітичного та біліарно-екскреторного синдромів, тому в плані лабораторного обстеження обов’язковим є ретельне виключення інфікування вірусами гепатитів (В, С, D, Е).
За нашими даними, в пацієнтів із СЖ лабораторні параметри корелювали між собою (рис.) із найпотужнішими центрами фокусування – лейкоцити та моноцити периферичної крові, ЛФ, АЛТ й індекс де Рітіса. Звертає на себе увагу те, що рівень моноцитів периферичної крові, які є основними антигенпрезентувальними клітинами, за умови класичного бактеріального запалення зі збільшенням лейкоцитозу, навпаки, зменшувався й був паралельним змінам ШОЕ. Тобто в пацієнтів із ДГБ будь-яке бактеріальне запалення перебігає атипово, свідченням чого також є відсутність інших маркерів запалення (С-реактивний білок, загальний фібриноген, серомукоїди, сіалові кислоти) в плеяді істотних кореляцій. Підтвердженням цього була поява аденопатії та лімфаденіту з нагноєнням атероми (3,2%) серед обстежених 63 пацієнтів із ДГБ. Натомість прогресування синдрому холестазу в пацієнтів із ДГБ асоціюється з цитолізом гепатоцитів і порушеннями вуглеводного метаболізму [6].
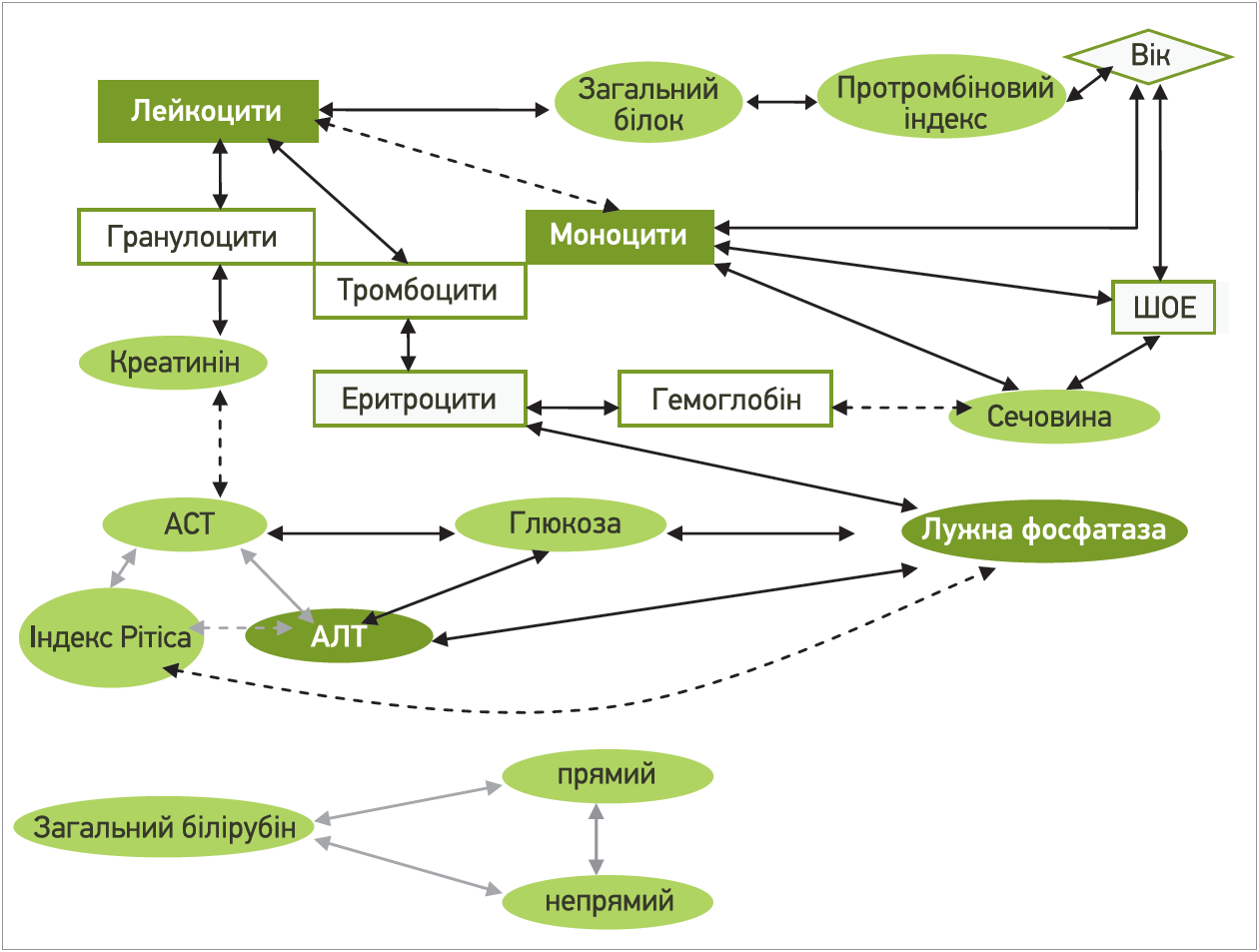
Рис. Істотні кореляційні зв’язки лабораторних параметрів у пацієнтів із СЖ
Найнесприятливішою ознакою для пацієнтів із СЖ виявилася лейкопенія периферичної крові, оскільки вона супроводжувалася (1) зменшенням гранулоцитів, яке асоціювалося зі зростанням трансаміназ, ЛФ і гіперглікемією; (2) тромбоцитопенією, що корелювала з анемію та зростанням сечовини як маркера підвищеного катаболізму; (3) моноцитозом, який також корелював зі вмістом сечовини та ШОЕ; (4) гіпопротеїнемією, що асоціювалася з гіпокоагуляцією (за протромбіновим індексом). Особливо небезпечними такі прояви стають в осіб старшого віку (істотні зв’язки з віком). Тобто лейкопенію периферичної крові у хворих на СЖ можна вважати простим інформативним маркером несприятливого клінічного перебігу й потреби перегляду лікувальної тактики. І навпаки: прогресування ураження печінки зі зростанням трансаміназ і ЛФ опосередковано призводитиме до послаблення антибактеріального захисту, геморагічного й анемічного синдромів, активації неспецифічного запалення. Хоча в наше дослідження були залучені молоді (середній вік – 29 років) військовослужбовці, середні значення лабораторних показників яких були в межах норми, у 3,2% із них загальна кількість лейкоцитів була низькою (<4×109/л), тобто саме на них варто звернути увагу лікарям [7].
Інструментальне дослідження печінки та жовчних шляхів у пацієнтів із СЖ може не виявляти змін або переважно давати картину підвищеної ехогенності печінки. Часто в таких пацієнтів сонографічно виявляються холелітіаз і висока кислотність шлункового соку (РН-метрія) із запальними чи ерозивно-виразковими змінами слизової оболонки стравоходу, шлунка, дванадцятипалої кишки (ФЕГДС). Обстеження 63 пацієнтів із СЖ показало, що тільки в 6,4% хворих на ДГБ була нормальна картина під час ФЕГДС. В інших виявлялися ураження стравоходу (гастроезофагеальна рефлюксна хвороба – 4,2%, езофагіт – 14,9%), шлунка (поліп без метаплазії – 2,1%, гастрит – 63,8%, ерозії шлунка – 10,6%, дуоденогастральний рефлюкс – 31,9%, гастропатія еритематозна – 2,1%) та 12-палої кишки (виразки – 10,6%, рубцева деформація – 12,8%, бульбіт і дуоденіт – 38,3%) [8].
Перебіг СЖ є хвилеподібним із періодами ремісії та загострення, що часто провокуються алкоголем, фізичним навантаженням, інтеркурентними хворобами. Зі зростанням віку пацієнтів збільшується частота непереносимості алкоголю, жирів, з’являється гепатомегалія, зростає активність АЛТ та ЛФ. СЖ у половині випадків поєднується з іншими хворобами, найчастіше з гепатитом, холелітіазом, хронічними запаленнями жовчних шляхів, гастритами, дуоденітами та пептичними виразками. За нашими даними, частота такого поєднання наближається до 100%.
Синдром Кріглера – Найяра успадковується за автосомно-рецесивним типом (дефектний ген зменшення кон’югаційної функції печінки) та характеризується майже повною нездатністю печінки кон’югувати білірубін унаслідок дефіциту глюкуронілтрансферази. Значна гіпербілірубінемія виникає з перших днів (синдром переважно діагностується в новонароджених) і триває все життя. Описано два типи: 1 – повна чи майже повна відсутність ферменту, несприятливий прогноз; 2 – описаний Arias у 1962 році (синдром Аріаса), зменшення активності ферменту <10%, що відповідає на лікування фенобарбіталом, прогноз сприятливіший. Диференційна діагностика передусім проводиться з фізіологічною та гемолітичною жовтяницями новонароджених. Особливостями синдрому Кріглера – Найяра є (1) ураження центральної нервової системи, в тому числі фатальні, коли під час секції виявляються жовте забарвлення та некроз ядер (kernicterus); (2) ахолічні випорожнення; (3) звичайна тривалість життя еритроцита; (4) постійно високий рівень білірубіну крові та (5) нормальна бромсульфалеїнова проба, яка свідчить про нормальну екскреторну здатність печінки, (6) у складі жовчі виявляються лише сліди білірубіну. Якщо такі пацієнти виживають, у них спостерігається відставання у фізичному та психічному розвитку. Прогноз загалом несприятливий.
Синдром Люсі – Дріскол – рідкісна родинна транзиторна гіпербілірубінемія новонароджених – успадковується за автосомно-рецесивним типом від матері, що має цю хворобу. Описано менше півсотні таких випадків, жовтяниця з’являється на 3-5-й день життя та зникає через 3 тиж. Окрім генетичного дефекту глюкуронілтрансферази, синдром також пов’язаний із наявністю в грудному молоці матері стероїдного фактора, який іще більше пригнічує фермент уридиндифосфат-глюкуронілтрансферазу, що призводить до вираженішого порушення кон’югації білірубіну, проте сам цей фактор дотепер не встановлений. Якщо такі пацієнти виживають, розвиток дитини не страждає, проте первинне зростання білірубіну може призводити до судом унаслідок уражень ядер центральної нервової системи (kernicterus).
Синдром Дабіна – Джонсона – кон’югована форма ДГБ – успадковується за автосомно-домінантним типом і посідає друге місце за частотою виявлення серед ДГБ після СЖ. Найчастіше трапляється серед іранських євреїв. У патогенезі відіграє роль порушення екскреторної функції печінки, що призводить до регургітації білірубіну; часто спостерігається порушення активності протромбіну. Синдром характеризується хронічною або інтермітувальною гіпербілірубінемією зі змінами проб екскреторної функції печінки. Морфологічно відрізняється тим, що в гепатоцитах накопичується темний пігмент (чорний колір печінки). Клінічні симптоми є неспецифічними: втомлюваність, біль у животі, нудота, втрата апетиту, збільшення печінки. Критерії діагнозу: повторне підвищення рівня бромсульфалеїну крові після навантаження ним, не контрастуються чи пізно контрастуються жовчні шляхи, темний (чорний) колір печінки при лапароскопії. Прогноз сприятливий.
Синдром Ротора – автосомно-рецесивно успадкована ДГБ, що найбільше подібна до синдрому Дабіна – Джонсона. Характеризується порушенням екскреторної функції печінки, є одночасно кон’югованою та некон’югованою ДГБ. Проявляється хронічною чи періодичною жовтяницею зі зростанням зв’язаного пігменту крові. При цьому синдромі часто спостерігається гепатоспленомегалія. На відміну від синдрому Дабіна – Джонсона, відзначається нормальна гістологія печінки, жовчні шляхи візуалізуються (повільніше чи слабше, ніж у нормі), а вміст копропорфірину сечі нормальний або <70% його складає ізомер‑1 (при синдромі Дабіна – Джонсона >80%). Не потребує лікування. Прогноз сприятливий.
Диференційна діагностика
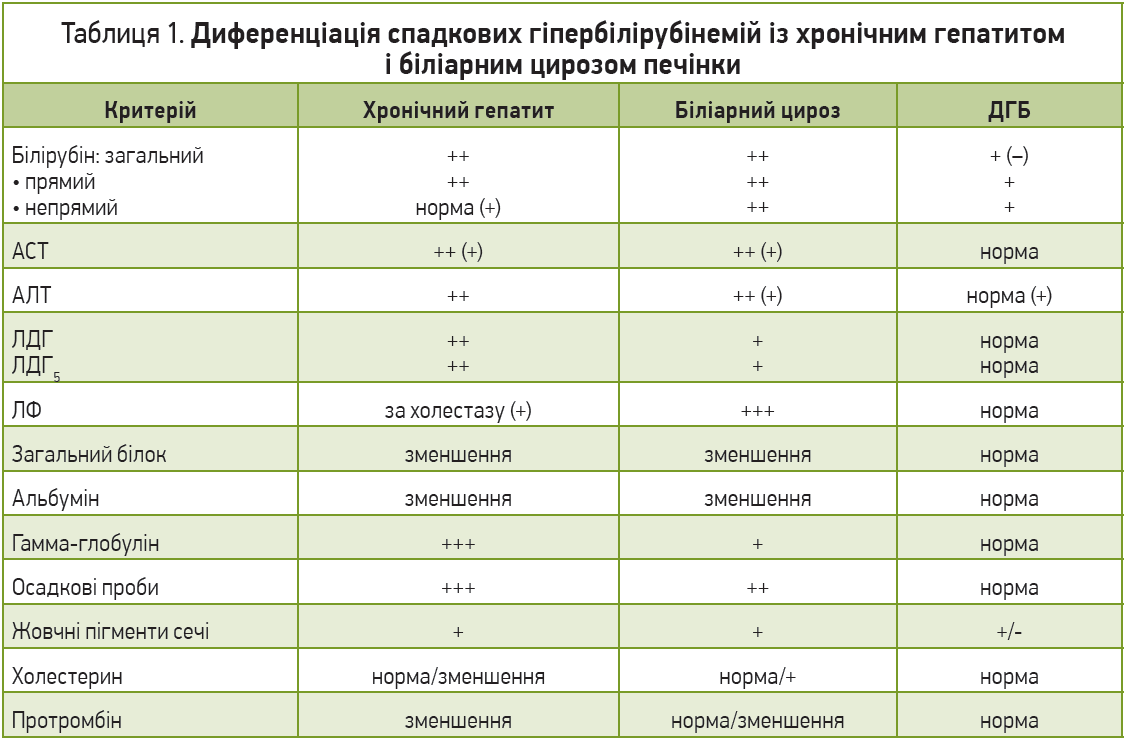
Диференційна діагностика всіх ДГБ проводиться, на жаль, лише методом виключення та є досить складною. Остаточно питання в багатьох випадках може бути вирішене лише після морфологічного дослідження біоптату печінки та генетичних досліджень. Слід пам’ятати про те, що наявність ДГБ у пацієнта часто слугує основою та провокувальним фактором розвитку інших уражень (вірусних, токсичних), тобто присутності відразу декількох механізмів і шляхів уражень. Найбільш важливою та складною є диференціація ДГБ з іншими хворобами печінки (табл. 1). Хронічні холестатичні жовтяниці (внутрішньо- та позапечінкові) характеризуються підвищенням зв’язаного білірубіну, симптомами холестазу, свербінням шкіри, високою активністю ЛФ, гіперхолестеринемією. Проте прицільна увага має бути спрямована на використання чутливіших, аніж скринінгові, методів діагностики вірусів гепатитів (полімеразно-ланцюгова реакція) та, можливо, маркерів автоімунних реакцій. Біліарні форми цирозів печінки проявляються зазвичай високими рівнями білірубіну, частіше трапляються в осіб середнього віку, але так само не мають патогномонічних ознак за винятком наявності антимітохондріальних антитіл за умови первинного біліарного цирозу. Для виключення вторинних біліарних цирозів потрібно прицільно визначити стан жовчовивідних шляхів, використовуючи не лише стандартне сонографічне дослідження, а й ретроградну панкреатохолангіографію. Наша клінічна практика свідчить, що присутність камінця в протоках може бути субклінічною (відсутність типового больового синдрому, незначна гіпербілірубінемія до 30 мкмоль/л, відсутність сонографічних проявів при стандартному обстеженні). Виключення алкогольних токсичних гепатитів потребує ретельного збору анамнезу та визначення вмісту γ-глутамілтранспептидази, яка вважається неспецифічним маркером алкогольного ураження. Слід пам’ятати, що пацієнти часто дають неправдиву інформацію про кількість ужитого алкоголю, особливо на стадіях збереження критичного ставлення до себе, тобто на початку алкогольних токсичних змін. Активація гемолізу при ДГБ потребує виключення гемолітичних анемій, для котрих притаманні (1) зменшення осмотичної та механічної резистентності еритроцитів, зміна їхньої морфології, підвищена екскреція уробіліногенових тіл; (2) накопичення заліза у вигляді гемосидерину в печінкових клітинах; (3) початок клінічних проявів до 10-річного віку хворого; (4) часто спленомегалія та виражена анемія, що рідко діагностується за умови гепатозів; (5) зменшення тривалості життя еритроцитів (менше 15 днів), тоді як у разі гепатозів вона хоч і зменшена, але перевищує 15 днів. Важливою є диференціація ДГБ між собою (табл. 2).
Лікування
Терапія ДГБ дотепер залишається проблемною, й багато питань є дискусійними, хоча не викликає сумнівів, що пацієнти мають перебувати під спостереженням лікаря. Правова база обстеження та лікування таких пацієнтів іще недостатньо оформлена. Регламентується лікування в дітей Наказом МОЗ № 255 від 27.04.2006. («Про затвердження клінічного протоколу надання неонатологічної допомоги дітям «Жовтяниці новонароджених»). Пацієнти з ДГБ мають обмеження щодо військової служби, які залежать, однак, від ступеня порушень функції печінки, що визначається індивідуально (Наказ МОЗ України № 402 від 14.08.2008 «Про затвердження Положення про військово-лікарську експертизу в Збройних Силах України»).
Лікування має обов’язково розпочинатися з модифікації способу життя, що потребує ретельної тривалої роботи та зусиль як пацієнта, так і лікаря. Протипоказані значні фізичні навантаження й алкоголь. Харчування має бути раціональним із контролем калоражу для запобігання збільшенню маси тіла та прогресуванню стеатозу печінки. Обов’язковою умовою є перегляд усіх медикаментів, які приймає пацієнт, і виключення засобів, які спричиняють або збільшують холестаз у печінці, призводять до некрозу гепатоцитів (табл. 3), лейкопенії. Дози всіх медикаментів мають бути ретельно вивірені, кількість лікарських засобів, які пацієнт приймає постійно чи тривало, має бути зменшена до мінімуму [9].

Виключення засобів, які спричиняють або збільшують холестаз і цитоліз у печінці чи призводять до лейкопенії, зумовлює певні труднощі. Проблемним стає лікування інших хвороб травної системи, які є супутніми в понад 90% пацієнтів із ДГБ. За даними Державного формуляра лікарських засобів, який містить рекомендації щодо раціонального призначення та використання препаратів з урахуванням ефективності, безпеки й економічної доцільності їх застосування, лейкопенію та холестаз одночасно можуть спричиняти всі макроліди, синтетичні пеніциліни, метронідазол, тетрацикліни й інгібітори протонної помпи, які становлять основу антигелікобактерної терапії, причому у високих дозах, а тривалість такої терапії збільшується з кожним оновленням Маастрихтського консенсусу. Цілком можливо, що саме антигелікобактерна й антикислотна терапії могли провокувати клінічні та лабораторні прояви ДГБ у більшості обстежених нами пацієнтів.
Також лейкопенію одночасно з холестазом можуть зумовлювати всі блокатори Н2-гістамінових рецепторів, практично всі антибактеріальні засоби, що використовуються нині в клінічній практиці, кардіологічні препарати (блокатори кальцієвих каналів, інгібітори ангіотензинперетворювального ферменту, сартани, сечогінні, в тому числі їх фіксовані комбінації, статини), антикоагулянти, деякі пероральні гіпоглікемічні (глімепірид), антипроліферативні (лефлуномід) і нестероїдні протизапальні препарати. Крім того, не варто призначати засоби, які спричиняють моноцитоз (гризеофульвін і галоперидол).
Оскільки етіотропного лікування генетичних дефектів не існує, основу терапії ДГБ становлять засоби умовно патогенетичного та симптоматичного спрямування: барбітурати, флумецинол, урсодезоксихолева кислота (УДХК), фототерапія, обмінні гемотрансфузії. Лише допоміжне значення мають інші гепатопротектори, вітаміни, жовчогінні та седативні засоби.
Механізми дії барбітуратів: 1) індукція утворення мікросомальних ферментів (глюкуронілтрансферази); 2) збільшення екскреції білірубіну; 3) збільшення утворення акцепторів білірубіну в гепатоциті; 4) активація утворення інших метаболітів гема. Фенобарбітал призначається в дозі 0,15 г на добу впродовж 1-2 міс. Флумецинол належить до групи гепатопротекторів, індукує оксидазну ферментативну активність мікросом печінки, посилює утворення глюкуронідів, сприяє виведенню з організму ендогенних та екзогенних метаболітів, збільшує виділення жовчі; на мікросомальні ферменти діє відносно вибірково, інших виражених фармакологічних ефектів не має; ефект з’являється через 5 днів застосування.
УДХК має антихолестатичний, холеретичний, цитопротекторний, антиапоптичний, імуномодулювальний, гіпохолестеринемічний, протизапальний, антиоксидантний, літолітичний і протипухлинний ефекти [10], що зумовлює доцільність її застосування за ДГБ. Власний досвід 10-річного спостереження свідчить про користь застосування УДХК у пацієнта із СЖ: загальний білірубін стабілізувався на рівні 105-110% від норми, а прямий білірубін знизився [11]; нині триває вивчення клінічної ефективності препаратів УДХК для лікування ДГБ у новонароджених. Фототерапія (лампи денного світла, кварцові лампи, сонячне опромінення) запропонована для лікування синдрому Кріглера – Найяра. Вона сприяє перетворенню білірубіну на полярніші діазонегативні, менш токсичні деривати.
Первинна профілактика ДГБ полягає лише в скринінгу населення на генетичні аномалії та вроджені хвороби, що на сучасному етапі розвитку охорони здоров’я в нашій країні нездійсненно. Проте загальна схема відповідає раніше описаним етапам гепатопротекції та профілактики уражень печінки: І – оцінка індивідуального сукупного ризику уражень печінки; ІІ – визначення ключових індивідуальних цілей профілактики уражень печінки; ІІІ – індивідуальний підбір немедикаментозних і фармакологічних методів лікування та планування заходів для зниження ризику уражень печінки; IV – моніторинг стану печінки й ефективності лікування, корекція профілактичних методів, кожен з яких здійснюється за трьома напрямами: вірусне/автоімунне, алкогольне, медикаментозне/токсичне [1].
Список літератури знаходиться в редакції.
Медична газета «Здоров’я України 21 сторіччя» № 10 (503), 2021 р.
