



Клінічний перебіг синдрому Хантера: час вирішує все
Мукополісахаридоз 2 типу (або синдром Хантера) – рідкісне аутосомно-рецесивне лізосомне захворювання, яке характеризується хронічним прогресуючим перебігом та ураженням багатьох органів і систем, зокрема опорно-рухового апарату, дихальної, серцево-судинної та нервової системи. Незважаючи на низьку поширеність, актуальність проблеми зумовлена тяжкістю клінічних проявів та відсутністю можливостей радикального лікування, що вимагає комплексного мультидисциплінарного підходу і своєчасного призначення патогенетичної терапії. Важливу роль у ранньому виявленні синдрому Хантера в дитячому і підлітковому віці відіграють сімейні лікарі та педіатри, яким варто обов’язково звернути увагу на характерну сукупність клінічних ознак (так звані червоні прапорці). Саме рання діагностика і скерування пацієнта до медичного генетика дозволяють своєчасно розпочати патогенетичну терапію та сповільнити прогресування інвалідизації при цьому захворюванні.
Мукополісахаридоз (МПС) 2 типу, відомий також як синдром Хантера (СХ), є рідкісним аутосомно-рецесивним захворюванням, вперше описаним доктором Чарльзом Хантером у 1917 році [1]. СХ спричинений дефіцитом лізосомального ферменту ідуронат-2-сульфатази (ІДС) [2, 3], який відповідає за розщеплення глікозаміногліканів, таких як дерматансульфат і гепарансульфат. Мутації в гені ІДС, розташованому в довгому плечі Х-хромосоми (Xq28), призводять до зниження активності або повної відсутності ферменту ІДС. Внаслідок цього прогресує накопичення глікозаміногліканів у лізосомах різних тканин, що спричиняє клінічні прояви захворювання.
Захворюваність на МПС 2 типу становить приблизно 1,3 випадки на 100 тис. новонароджених чоловічої статі [4]. Оскільки ген ІДС знаходиться в Х-хромосомі, СХ, як правило, маніфестує у чоловіків, проте описані поодинокі випадки захворювання й у жінок з аномальним набором Х-хромосом [5, 6].
СХ є прогресуючим, багатосистемним, хронічним і загрозливим для життя захворюванням зі значною мінливістю тяжкості симптомів, швидкості прогресування і віку початку у різних пацієнтів. Хоча хвороба традиційно класифікується як легка чи тяжка залежно від тяжкості проявів і наявності чи відсутності ураження центральної нервової системи (ЦНС), СХ слід розглядати як континуум клінічних фенотипів – від досить м’яких до надзвичайно тяжких. Існує значна варіабельність вираженості симптомів, віку маніфестації та швидкості прогресування у різних пацієнтів. Перехід від однієї «крайньої» форми до іншої є поступовим. Тобто поняття «континуум між двома крайніми формами» означає, що СХ має широкий спектр можливих варіантів перебігу – від відносно легкого (повільна прогресія, переважно соматичні прояви, нормальний інтелект) до надзвичайно тяжкого (швидке наростання симптомів, рання інвалідизація, летальність). Між цими полюсами існують численні проміжні форми різного ступеня тяжкості [7, 8].
Пацієнти з СХ зазвичай народжуються без жодних симптомів та ознак захворювання. Проте з часом, зазвичай між 1,5 та 4 роками, у них починають з’являтися певні прояви, які поступово прогресують [9]. Спочатку це можуть бути неспецифічні скарги на часті респіраторні інфекції, проблеми зі слухом або затримка мовного і психомоторного розвитку. Проте згодом формується характерна клінічна картина. Важливими червоними прапорцями, що мають насторожити лікаря і стати приводом для подальшого дообстеження з метою виключення СХ, є:
- прогресуюча дисморфія обличчя з грубими рисами, широким носом, великими губами, виступаючим лобом;
- відставання у фізичному та розумовому розвитку;
- множинні контрактури суглобів та обмеження рухливості;
- низький зріст для вікової норми;
- збільшення розмірів печінки і селезінки;
- порушення з боку серцево-судинної системи;
- рецидивуючі респіраторні інфекції на тлі порушень легеневої функції (рис. 1) [10, 11].
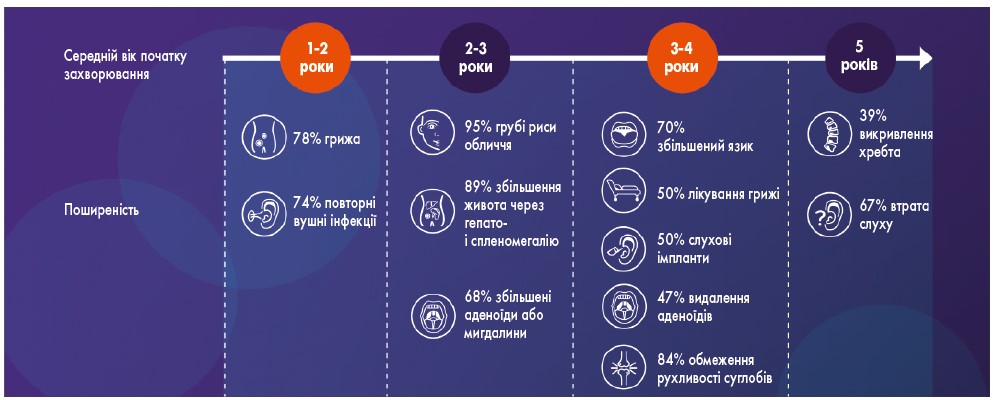 Рис. 1. Клінічні прояви СХ
Рис. 1. Клінічні прояви СХ
Наявність комбінації декількох із зазначених проявів має стати приводом для невідкладного звернення до генетика і проведення лабораторної діагностики (визначення рівня глікозаміногліканів, активності лізосомних ферментів, молекулярно-генетичного дослідження) для верифікації або спростування діагнозу СХ. Адже лише своєчасна діагностика і лікування дасть змогу уповільнити прогресування захворювання і покращити якість життя дитини (рис. 2).
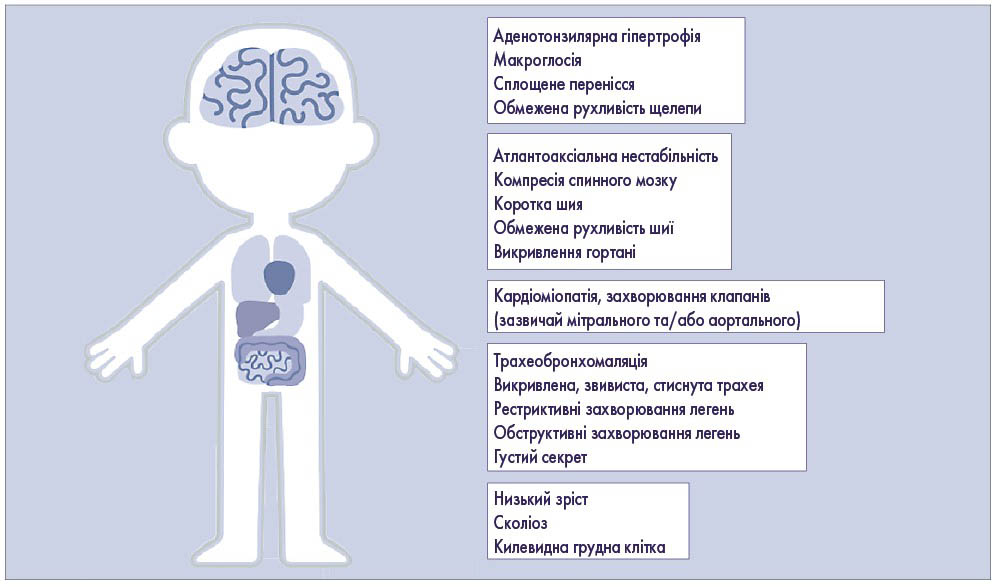 Рис. 2. Дитина з мукополісахаридозом має низький зріст, акроцефальну голову, грубі риси обличчя та вдавлене перенісся
Рис. 2. Дитина з мукополісахаридозом має низький зріст, акроцефальну голову, грубі риси обличчя та вдавлене перенісся
Клінічний випадок 1*
Пацієнт чоловічої статі віком 3 роки був госпіталізований з респіраторними симптомами на тлі анамнезу рецидивуючих інфекцій дихальних шляхів. З акушерського анамнезу відомо, що вагітність ускладнилася гестаційним ЦД і полігідрамніоном. Після народження дитину консультували щодо аномалій зовнішності, проте батьки відмовилися від обстеження. Згодом у віці 6 місяців була діагностована дисплазія кульшових суглобів, а також відзначалося відставання у фізичному і мовному розвитку. Об’єктивно при огляді було виявлено характерні для СХ фенотипові ознаки.
Для уточнення діагнозу було здійснено комплексне лабораторне та інструментальне обстеження пацієнта. Зокрема, проведений аналіз активності основних лізосомальних ферментів методом мас-спектрометрії виявив різке зниження рівня ІДС (0,05 мкмоль/л/год при референтному діапазоні 2,0-20) на тлі нормальних показників інших досліджуваних ферментів (α-ідуронідази, арилсульфатази B, галактозо-6-сульфатази та ін.). Ці прояви стали приводом для подальшого генетичного обстеження, за результатами якого був встановлений діагноз МПС 2 типу (СХ), що підтверджувалося низьким рівнем активності ферменту ІДС.
Отже, фенотипові ознаки у поєднанні з ураженням життєво важливих органів стали тими червоними прапорцями, котрі сприяли своєчасній діагностиці спадкового захворювання та визначення подальшого менеджменту [12].
Клінічний випадок 2
Пацієнт 2 років потрапив до медичного закладу зі скаргами на рецидивуючі інфекції верхніх дихальних шляхів. Об’єктивно: грубі риси обличчя зі збільшеним черепом, гепатоспленомегалія, пупкова та пахова грижі, контрактури суглобів. У пацієнта спостерігалася психомоторна затримка (загальний коефіцієнт інтелекту 79 на основі шкали WPPSI-III), гіперактивність і проблеми з увагою. Об’єктивно при огляді: зріст 85 см (<3 центиля), вага 12 кг (<3 центиля). Спостерігається грубий дисморфізм обличчя: велика голова з виступаючим лобом, гіпертелоризм очей, плоска спинка носа, товсті губи. Деформація грудної клітки. Аускультативно – дихання жорстке, хрипи не вислуховуються. ЧД 20/хв. Тони серця глухі, акцент ІІ тону над легеневою артерією. Живіт збільшений за рахунок гепатоспленомегалії. Печінка +4 см, щільна. Селезінка +2 см. Статико-моторні функції не відповідають віку: дитина не ходить, мовлення відсутнє.
Незважаючи на виражену симптоматику, діагноз було встановлено лише у 4 роки на підставі комплексного клініко-лабораторного та інструментального обстеження, що виявило ознаки мультисистемного ураження і підвищення рівня глікозаміногліканів у сечі. Молекулярно-генетичний аналіз підтвердив гомозиготну мутацію в гені ІДС, асоційовану з тяжким фенотипом СХ. Було розпочато патогенетичну ферментозамісну терапію ІДС.
Отже, наявність у 2-річного пацієнта чоловічої статі комплексу симптомів у вигляді рецидивуючих респіраторних інфекцій, краніофаціального дисморфізму, множинних скелетних аномалій, гепатоспленомегалії та затримки психомоторного розвитку стало вектором менеджменту для обстеження в напрямку лізосомних хвороб накопичення, зокрема МПС. Комплексна діагностика із застосуванням лабораторних та інструментальних методів дозволила виявити маркери МПС 2 типу і верифікувати діагноз СХ у 4-річному віці з подальшим призначенням патогенетичної ферментозамісної терапії (ФЗТ) ІДС [13].
Сучасні стратегії лікування: минуле, сьогодення та майбутнє
За останні десятиліття відбувся значний прогрес у лікуванні спадкових лізосомних захворювань, зокрема МПС 2 типу. Сьогодні двома доступними стратегіями цільового лікування є ФЗТ і трансплантація гемопоетичних стовбурових клітин (ТГСК).
На сьогодні основним методом патогенетичного лікування СХ є ФЗТ – регулярні внутрішньовенні інфузії рекомбінантної ІДС [1]. Впровадження ФЗТ значно змінило природний перебіг захворювання і прогноз для пацієнтів, зокрема її застосування супроводжується покращенням пересування, легеневої функції та виживання [14-16]. Безпека й ефективність ФЗТ ІДС була продемонстрована у трьох клінічних дослідженнях: двох рандомізованих плацебо-контрольованих клінічних дослідженнях (TKT008 та TKT024) за участю дорослих і дітей віком старше 5 років і одному відкритому дослідженні безпеки (HGT-ELA-038) у дітей віком від 16 місяців до 7,5 року [17-19].
Експертний консенсус Американського коледжу медичної генетики та геноміки (American College of Medical Genetics and Genomics) 2020 р. рекомендував розпочати прийом ІДС під час встановлення діагнозу і до появи симптомів в осіб, у яких за генотипом прогнозується тяжкий перебіг СХ, та усім, хто має симптоми, зумовлені СХ, незалежно від прогнозованого ступеня тяжкості [20]. Ці рекомендації також відображені і в інших літературних джерелах [21]; зокрема, дані сучасних досліджень свідчать, що регулярні інфузії ІДС дозволяють ефективно контролювати більшість проявів СХ: зменшувати рівень глікозаміногліканів, покращувати функцію легень та опорно-рухового апарату, стабілізувати прогресування неврологічних розладів [22].
ТГСК є альтернативним методом лікування СХ, що базується на трансплантації донорських клітин кісткового мозку чи кордової крові, які здатні продукувати фермент ІДС і мігрувати в різні органи, включаючи ЦНС [23]. Потенційною перевагою ТГСК вважають одноразовість процедури на відміну від тривалої багаторазової ФЗТ [24, 25]. Проте аналіз результатів клінічних досліджень свідчить про відносно високу післятрансплантаційну смертність (до 8%), а також недостатню ефективність ТГСК щодо неврологічних симптомів СХ [26].
Таким чином, ФЗТ рекомбінантною ІДС є основним методом етіопатогенетичного лікування СХ, що базується на регулярних внутрішньовенних інфузіях ферменту для компенсації його дефіциту в організмі. Відповідно до чинних міжнародних рекомендацій, стандартна схема введення ІДС становить 0,5 мг/кг 1 раз/тиждень внутрішньовенно. Доведено, що такий режим ФЗТ асоціюється зі збільшенням виживаності пацієнтів, а також забезпечує регрес багатьох клінічних симптомів, зокрема зменшення контрактур суглобів, помірну редукцію розмірів печінки і селезінки, покращення функції зовнішнього дихання, стабілізацію гіпертрофії міокарда [27-30].
Збільшення досвіду застосування цієї терапії дозволило розширити її показання для педіатричних пацієнтів, включаючи немовлят з 6-місячного віку [31]. Зокрема, результати багатоцентрового клінічного дослідження HGT-ELA-038 (NCT00607386) продемонстрували порівнянну ефективність і безпеку ФЗТ ІДС у дітей із СХ віком від 16 місяців до 7,5 року. Отримані результати продемонстрували, що застосування ІДС забезпечило майже 60% зниження рівня глікозаміногліканів у сечі і зменшення розмірів печінки й селезінки [17]. Постфактум аналіз аспектів імуногенності також не виявив підстав для розгляду змін у веденні пацієнтів цієї групи [32].
Висновок
МПС 2 типу (або СХ) є рідкісним спадковим захворюванням з аутосомно-рецесивним типом успадкування, в основі якого лежить дефіцит лізосомального ферменту ІДС. Це призводить до прогресуючого накопичення глікозаміногліканів у тканинах і розвитку поліорганних порушень. Перші симптоми СХ зазвичай проявляються у ранньому дитинстві та можуть призвести до інвалідизації пацієнтів, якщо не розпочато своєчасне патогенетичне лікування.
Вирішальну роль у виявленні комплексу характерних червоних прапорців, що можуть вказувати на СХ, відіграють педіатри і сімейні лікарі. Саме рання діагностика та скерування до медичного генетика дають змогу призначити своєчасну ФЗТ, яка є основою етіопатогенетичного лікування цього захворювання відповідно до сучасних рекомендацій. Результати клінічних досліджень підтверджують, що завдяки комплексному мультидисциплінарному підходу можна досягти контролю над клінічною симптоматикою та уповільнити прогресування інвалідизації пацієнтів із СХ. Довготривала ФЗТ у комплексі з іншими підходами (хірургічні втручання, фізіотерапія, гідротерапія та симптоматична терапія) спрямована на зниження вмісту глікозаміногліканів у біологічних рідинах, регресію гепатоспленомегалії, покращення функції зовнішнього дихання і толерантності до фізичних навантажень, уповільнення прогресування скелетних деформацій (рис. 3) [33].
 Рис. 3. Терапевтична стратегія ведення СХ [19]
Рис. 3. Терапевтична стратегія ведення СХ [19]
Література
- Hunter C. A rare disease in two brothers. Proc R Soc Med. 1917. 10: 104-106.
- Sestito S., Ceravolo F., Grisolia M. et al. Profile of idursulfase for the treatment of Hunter syndrome. Research and Reports in Endocrine Disorders. 2015. 5: 79-90. https://doi.org/10.2147/RRED.S64347.
- Neufeld E.F., Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York. 2001. McGraw-Hill; 3421-3452.
- Beck M., Wijburg F.A., Gal A. Clinical utility gene card for: mucopolysaccharidosis type II. Eur J Hum Genet. 2012. 20 (1): 1.
- Tuschl K., Gal A., Paschke E. et al. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol. 2005. 32: 270-272.
- Pinto L.L., Vieira T.A., Giugliani R. et al. Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis. 2010. 5: 14.
- Martin R., Beck M., Eng C. et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008. 121: e377-e386.
- Wraith J.E., Beck M., Giugliani R. et al. Initial report from the Hunter outcome survey. Genet Med. 2008. 10: 508-516.
- Schwartz I.V., Ribeiro M.G., Mota J.G. et al. A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr Suppl. 2007. 96 (455): 63-70.
- Wraith J.E., Scarpa M., Beck M. et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008. 167: 267-277.
- Jones S.A., Almassy Z., Beck M. et al. HOS Investigators. Mortality and cause of death in mucopolysaccharidosis type II – a historical review based on data from the Hunter outcome survey (HOS). J Inherit Metab Dis. 2009. 32: 534-543.
- Al-Mashakbeh Y., Heissat N., Al-Shaibei A. et al. Congenital Diaphragmatic Hernia as a Presentation of Mucopolysaccharidosis in a 3-year-old child: A Case Report. Med J Islam Repub Iran. 2022 Oct 24. 36: 123. doi: 10.47176/mjiri.36.123.
- Gragnaniello V., Carraro S., Rubert L. et al. A new strategy of desensitization in mucopolysaccharidosis type II disease treated with idursulfase therapy: A case report and review of the literature. Molecular genetics and metabolism reports, 31, 100878. 2022. https://doi.org/10.1016/j.ymgmr.
- Burton B.K., Jego V., Mikl J. et al. Survival in idursulfase-treated and untreated patients with mucopolysaccharidosis type II: data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis. Nov. 2017. 40 (6): 867-874. doi: 10.1007/s10545-017-0075-x.
- Muenzer J., Gucsavas-Calikoglu M., McCandless S.E. et al. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab. Mar 2007. 90 (3): 329-37. doi: 10.1016/j.ymgme.2006.09.001.
- Muenzer J., Wraith J.E., Beck M. et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. Aug. 2006. 8 (8): 465-73. doi: 10.1097/01.gim.0000232477.37660.fb.
- Whiteman D.A., Kimura A. Development of idursulfase therapy for mucopolysaccharidosis type II (Hunter syndrome): the past, the present and the future. Drug Des Devel Ther. Aug 23. 2017. 11: 2467-2480. doi: 10.2147/DDDT.S139601.
- Puckett Y., Mallorga-Hernandez A., Montao A.M. Epidemiology of mucopolysaccharidoses (MPS) in United States: challenges and opportunities. Orphanet J Rare Dis. May 29. 2021. 16 (1): 241. doi: 10.1186/s13023-021-01880-8.
- Nan H., Park C., Maeng S. Mucopolysaccharidoses I and II: Brief Review of Therapeutic Options and Supportive/Palliative Therapies. Biomed Res Int. Dec 4. 2020: 2408402. doi: 10.1155/2020/2408402.
- McBride K.L., Berry S.A., Braverman N. ACMG Therapeutics Committee. Treatment of mucopolysaccharidosis type II (Hunter syndrome): a Delphi derived practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. Nov. 2020. 22 (11): 1735-1742. doi: 10.1038/s41436-020-0909-z.
- Ream M.A., Lam W.K.K., Grosse S.D. et al. Evidence and recommendation for mucopolysaccharidosis type II newborn screening in the United States. Genet Med. 2023 Feb. 25 (2): 100330. doi: 10.1016/j.gim.2022.10.012.
- Yee K.S., Alexanderian D., Feng Y. et al. Impact of the Timing of Enzyme Replacement Therapy Initiation and Cognitive Impairment Status on Outcomes for Patients with Mucopolysaccharidosis II (MPS II) in the United States: A Retrospective Chart Review. J Health Econ Outcomes Res. Aug 29. 2022. 9 (2): 67-76. doi: 10.36469/001c.36540.
- Araya K., Sakai N., Mohri I. et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol Genet Metab. Nov. 2009. 98 (3): 255-63. doi: 10.1016/j.ymgme.2009.05.006.
- Taylor M., Khan S., Stapleton M. et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol Blood Marrow Transplant. Jul. 2019. 25 (7): e226-e246. doi: 10.1016/j.bbmt.2019.02.012.
- Tanaka A., Okuyama T. et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: a nationwide survey in Japan. Mol Genet Metab. Nov. 2012. 107 (3): 513-20. doi: 10.1016/j.ymgme.2012.09.004.
- Guffon N., Bertrand Y., Forest I. et al. Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr. May. 2019. 154 (5): 733-7. doi: 10.1016/j.jpeds.2008.11.041.
- Pollard L.M., Jones J.R., Wood T.C. Molecular characterization of 355 mucopolysaccharidosis patients reveals 104 novel mutations. J Inherit Metab Dis. Mar. 2013. 36 (2): 179-87. doi: 10.1007/s10545-012-9533-7.
- Stapleton M., Kubaski F., Mason R.W. et al. Presentation and Treatments for Mucopolysaccharidosis Type II (MPS II; Hunter Syndrome). Expert Opin Orphan Drugs. 2017. 5 (4): 295-307. doi: 10.1080/21678707.2017.1296761.
- Grant N., Sohn Y.B., Ellinwood N.M. et al. Timing is everything: Clinical courses of Hunter syndrome associated with age at initiation of therapy in a sibling pair. Mol Genet Metab Rep. Feb 2. 2022. 30: 100845. doi: 10.1016/j.ymgmr.2022.100845.
- Tomita K., Okamoto S., Seto T. et al. Divergent developmental trajectories in two siblings with neuropathic mucopolysaccharidosis type II (Hunter sy drome) receiving conventional and novel enzyme replacement therapies: A case report. JIMD Rep. Jul 27. 2022. 62 (1): 9-14. doi: 10.1002/jmd2.12239.
- Lampe C., Atherton A., Burton B.K. et al. Enzyme replacement therapy in mucopolysaccharidosis II patients under 1 year of age. JIMD Rep. 2014. 14: 99-113.
- Pano A., Barbier A.J., Bielefeld B. Immunogenicity of idursulfase and clinical outcomes in very young patients (16 months to 7,5 years) with mucopolysaccharidosis II (Hunter syndrome). Orphanet J Rare Dis. 2015. 10: 50.
- Сelik B., Tomatsu S.C., Tomatsu S. et al. Epidemiology of Mucopolysaccharidoses Update. Diagnostics (Basel). Feb 10. 2021. 11 (2): 273. doi: 10.3390/diagnostics11020273.
Підготувала Анна Сочнєва
Матеріал створений за підтримки компанії «Такеда». Містить рекламу.
© ТОВ «Такеда Україна». Всі права захищені.
«ТАКЕДА» та ![]() є зареєстрованими торговельними марками компанії Takeda Pharmaceutical Company Limited
є зареєстрованими торговельними марками компанії Takeda Pharmaceutical Company Limited
VV-MEDMAT-100539
*Клінічні випадки включають реальний досвід пацієнтів із СХ і містять інформацію про клінічний досвід медичних працівників. Індивідуальний досвід може відрізнятися.
Тематичний номер «Педіатрія» № 1 (72) 2024 р.
СТАТТІ ЗА ТЕМОЮ Педіатрія
Вроджена дисфункція кори надниркових залоз (ВДКНЗ) – це захворювання з автосомно-рецесивним типом успадкування, в основі якого лежить дефект чи дефіцит ферментів або транспортних білків, що беруть участь у біосинтезі кортизолу. Рання діагностика і початок лікування пацієнтів з ВДКНЗ сприяє покращенню показників виживаності та якості життя пацієнтів....
Алергічний риніт (АР) є поширеним запальним захворюванням верхніх дихальних шляхів (ВДШ), особливо серед педіатричних пацієнтів. Ця патологія може знижувати якість життя, погіршувати сон та щоденну продуктивність. Метою наведеного огляду є надання оновленої інформації щодо епідеміології АР та його діагностики, з урахуванням зв’язку з бронхіальною астмою (БА). ...
Американська академія педіатрії (AAP) оновила рекомендації щодо контролю грипу серед дитячого населення під час сезону 2023-2024 рр. Згідно з оновленим керівництвом, для профілактики та лікування грипу в дітей необхідно проводити планову вакцинацію з 6-місячного віку, а також своєчасно застосовувати противірусні препарати за наявності показань. ...
Поширеність і вплив алергічних захворювань часто недооцінюють [1]. Ключовим фактором алергічної відповіді є імуноглобулін (Ig) Е, присутній на поверхні тучних клітин і базофілів. Взаємодія алергену з IgЕ та його рецепторним комплексом призводить до активації цих клітин і вивільнення речовин, у тому числі гістаміну, які викликають симптоми алергії [2]. Враховуючи ключову роль гістаміну в розвитку алергічних реакцій, при багатьох алергічних станах, включаючи алергічний риніт і кропив’янку, пацієнту призначають антигістамінні препарати [3, 4]....